The Contribution of MicroRNAs to the Inflammatory and Neoplastic Characteristics of Erdheim–Chester Disease

 , , ,
, , ,
Simple Summary
Abstract
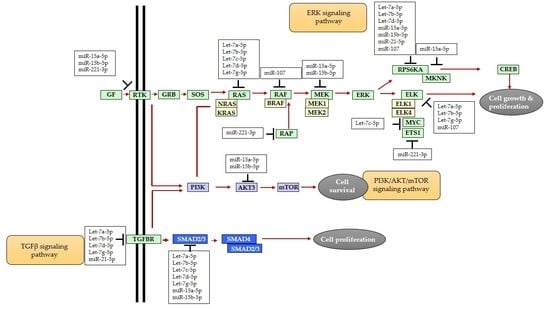
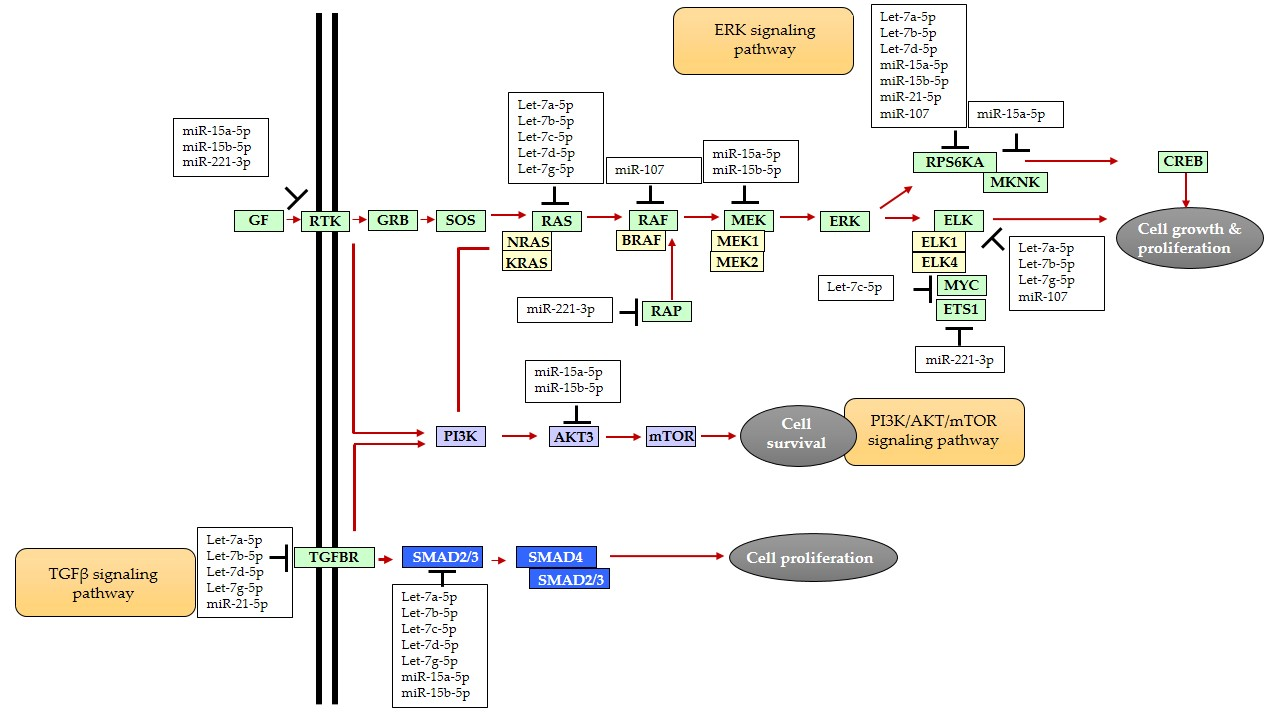
1. Introduction
2. Results
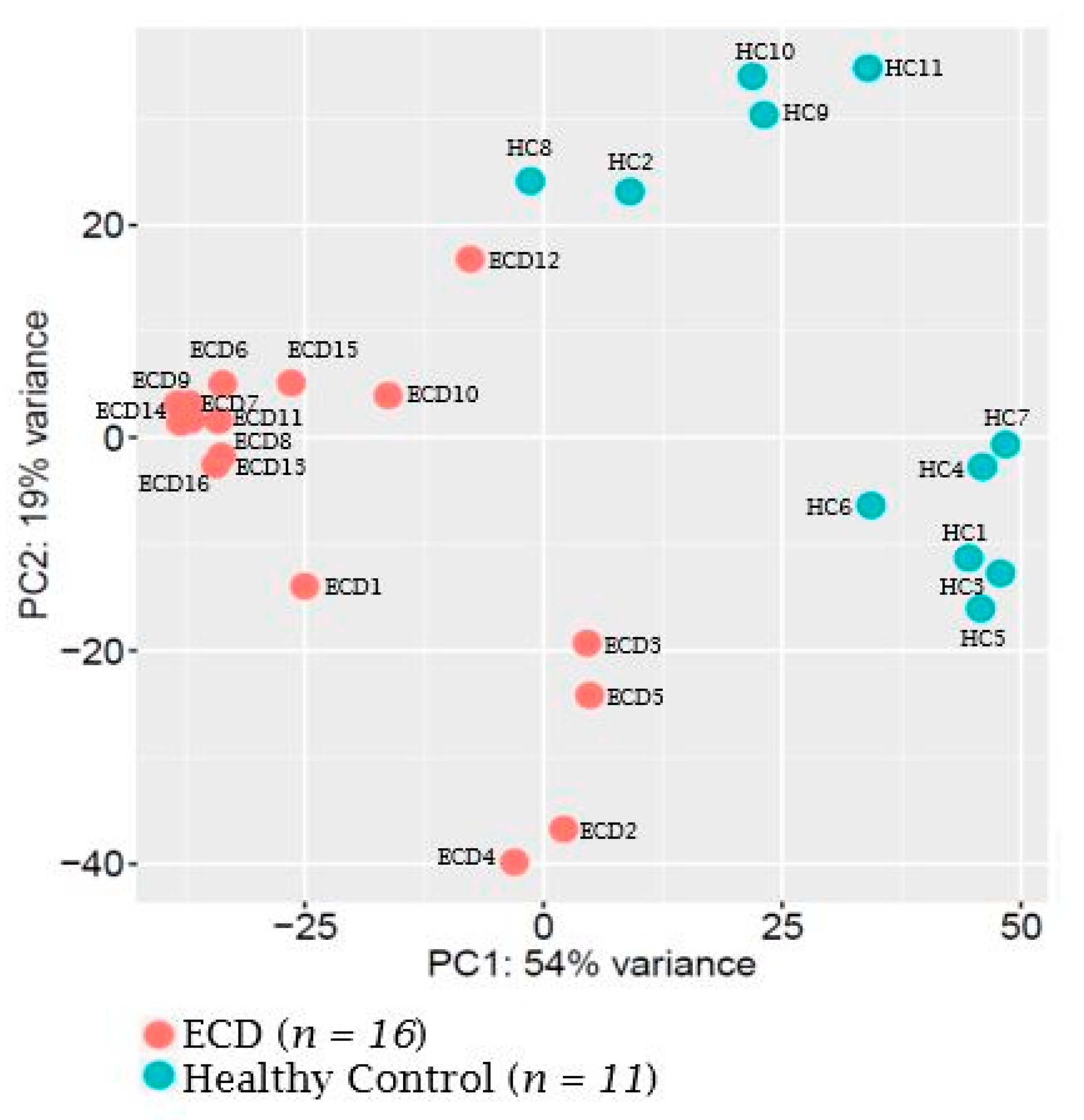
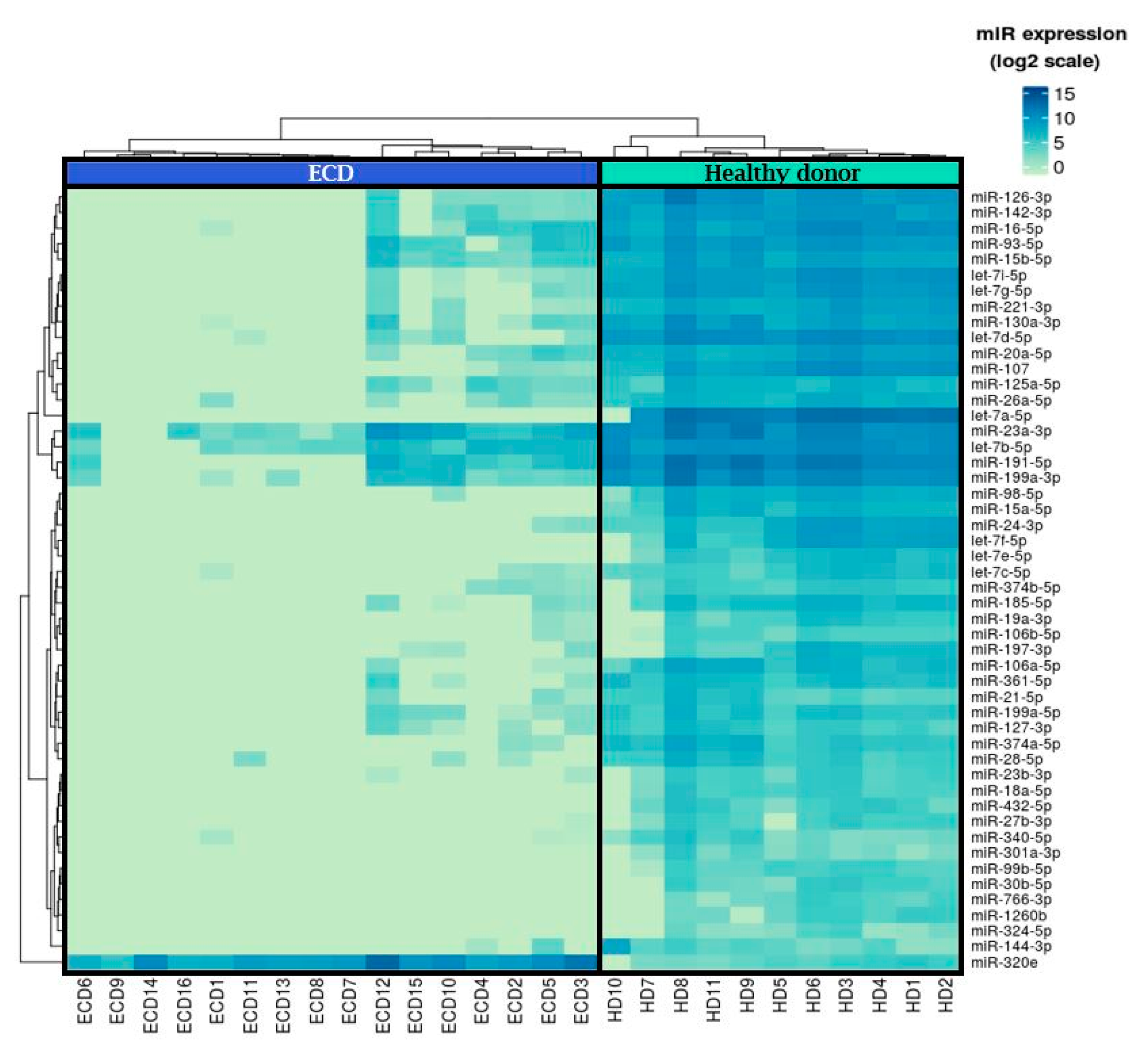
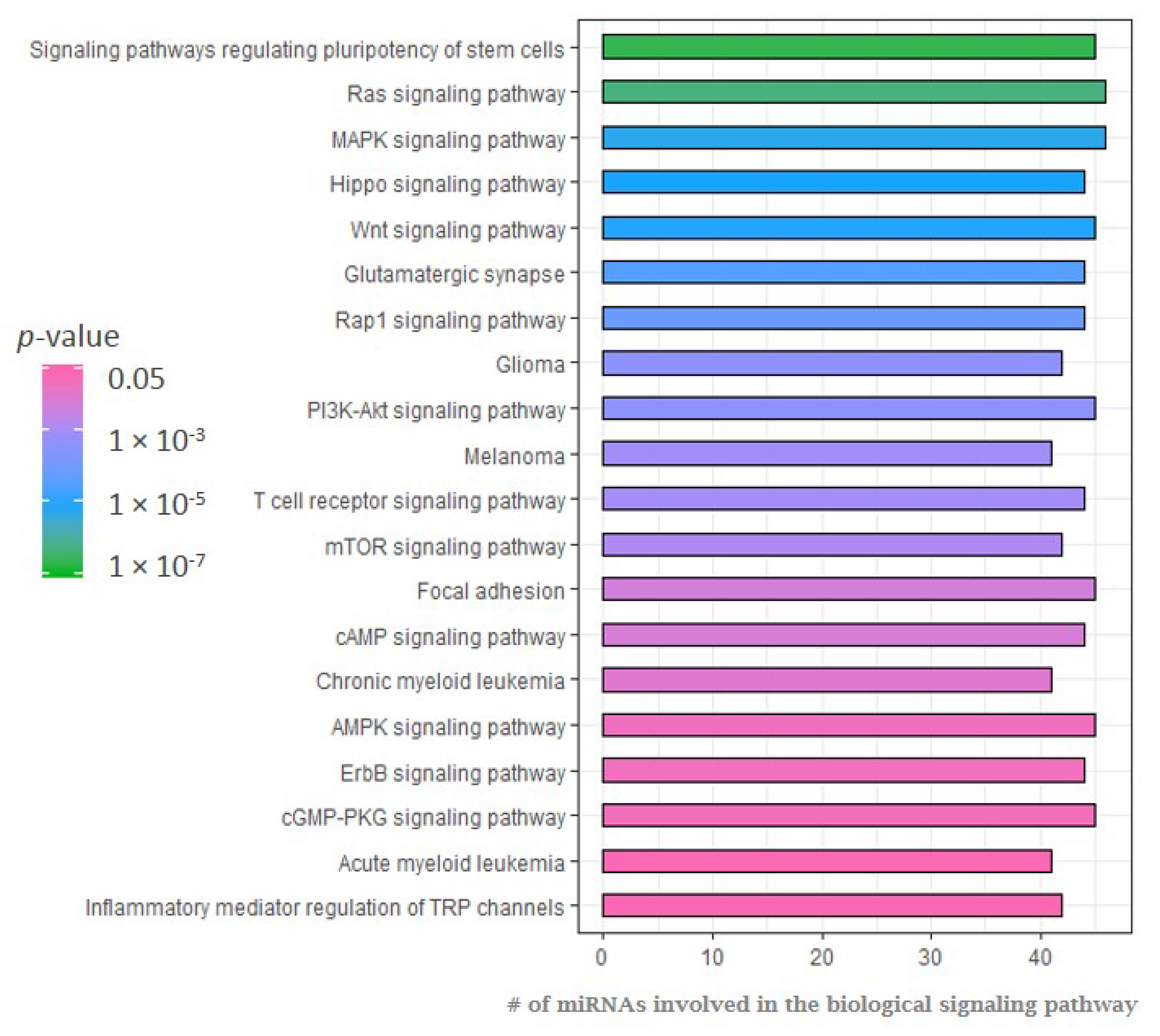
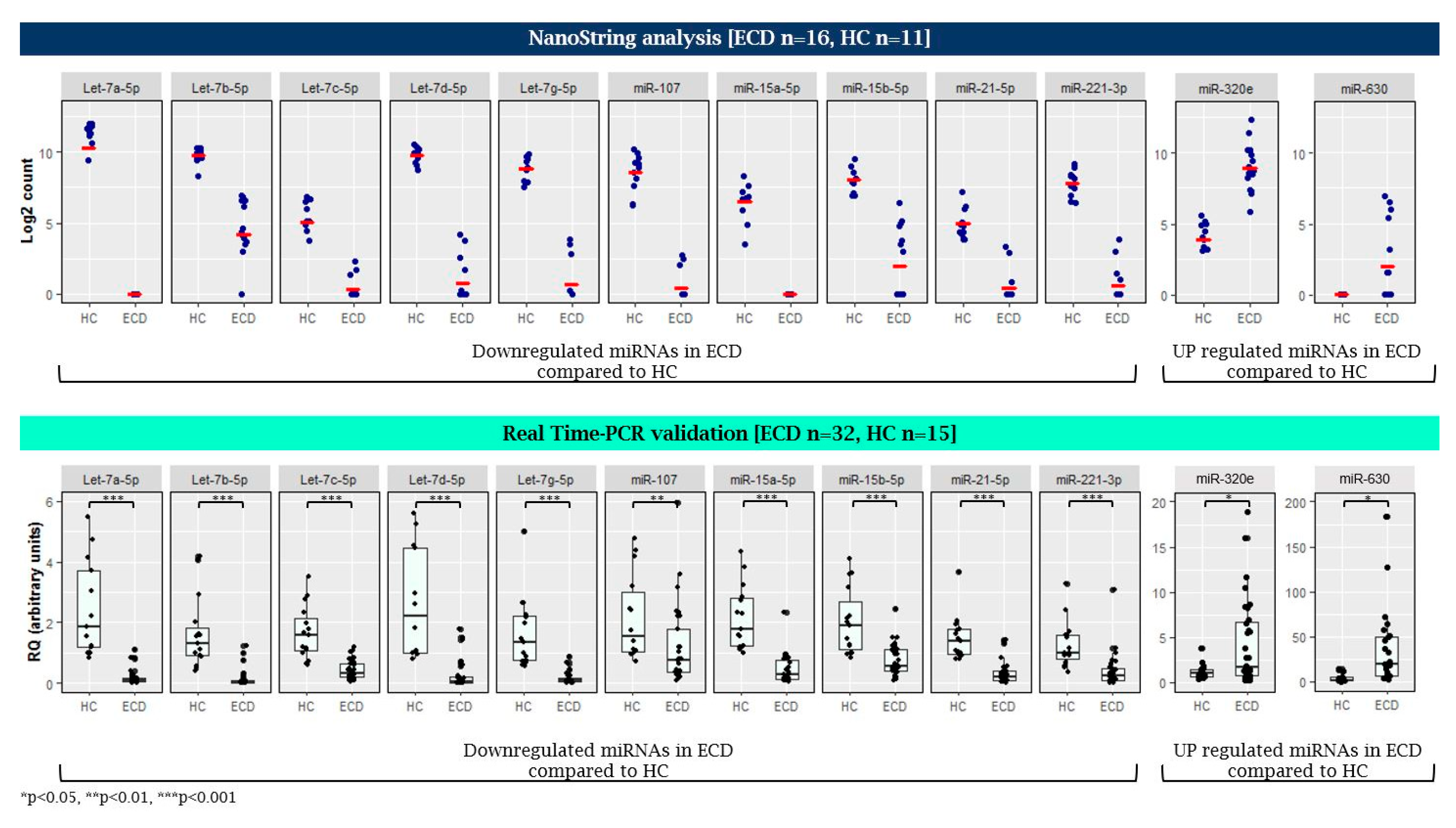
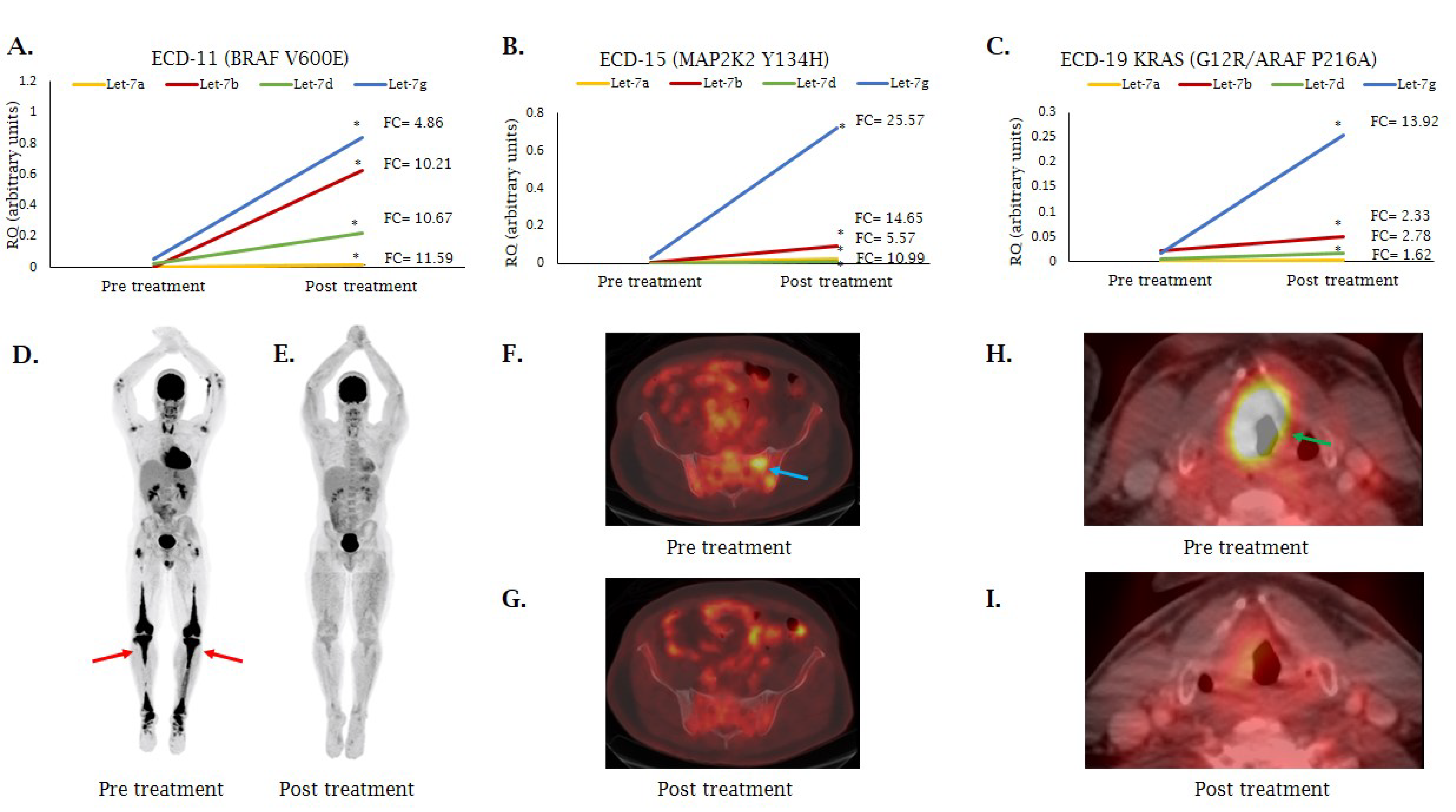
2.1. MicroRNA Expression Patterns in Erdheim–Chester Disease (ECD) Patients as Compared with Healthy Individuals
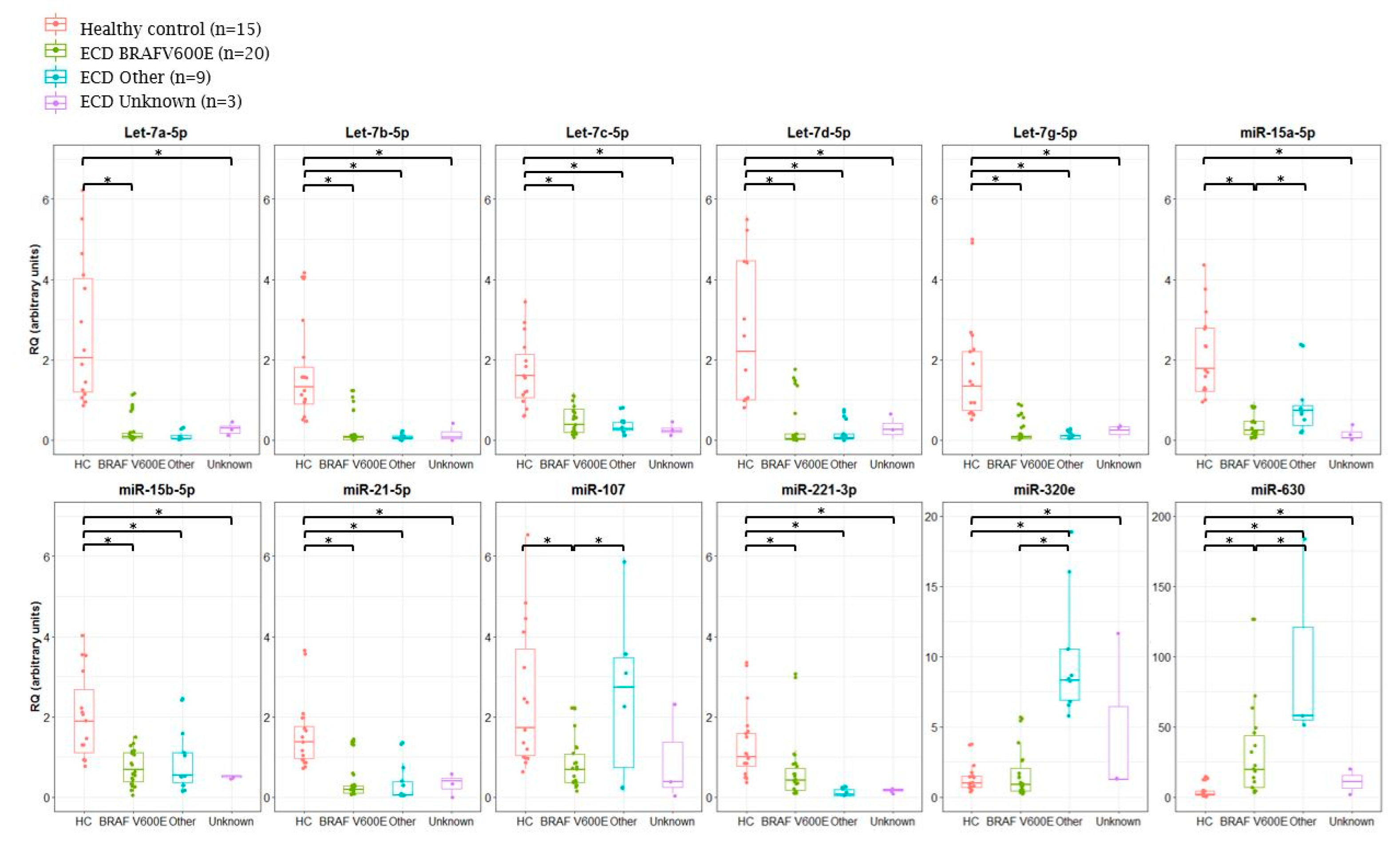
2.2. Analysis of miRNA Expression in ECD Patients by Mutation Type
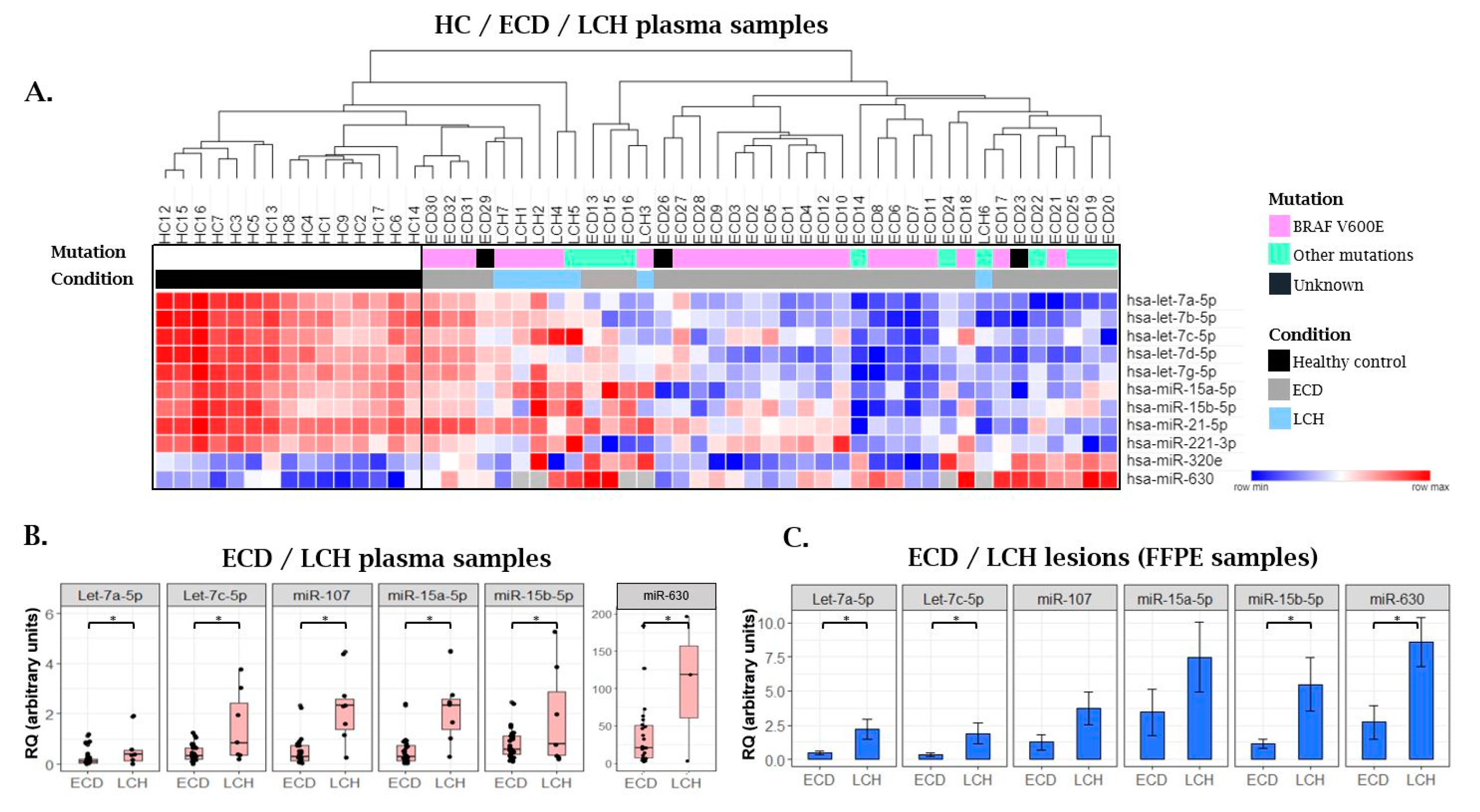
2.3. MiRNA Expression Patterns in Langerhans Cell Histiocytosis (LCH) Patients as Compared with ECD Patients and Healthy Individuals
2.4. MiRNA Expression Following MAP/-ERK Signaling Cascade Inhibition
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Genomic Analyses
4.3. MiRNA Purification
4.4. NanoString Analysis
4.5. Quantitative Real-Time PCR (qRT-PCR) Validation
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Goyal, G.; Heaney, M.L.; Collin, M.; Aubart, F.C.; Vaglio, A.; Durham, B.H.; Hershkovitz-Rokah, O.; Girschikofsky, M.; Jacobsen, E.D.; Toyama, K.; et al. Erdheim-Chester disease: Consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood 2020, 135, 1929–1945. [Google Scholar] [CrossRef] [PubMed]
- Dagna, L.; Corti, A.; Langheim, S.; Guglielmi, B.; De Cobelli, F.; Doglioni, C.; Fragasso, G.; Sabbadini, M.G.; Ferrarini, M. Tumor Necrosis Factor α As a Master Regulator of Inflammation in Erdheim-Chester Disease: Rationale for the Treatment of Patients With Infliximab. J. Clin. Oncol. 2012, 30, e286–e290. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Gorochov, G.; Charlotte, F.; Lvovschi, V.; Parizot, C.; Larsen, M.; Ghillani-Dalbin, P.; Hervier, B.; Kahn, J.-E.; Deback, C.; et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: A single-center series of 37 patients. Blood 2011, 117, 2783–2790. [Google Scholar] [CrossRef] [PubMed]
- Tomelleri, A.; Cavalli, G.; De Luca, G.; Campochiaro, C.; D’Aliberti, T.; Tresoldi, M.; Dagna, L. Treating Heart Inflammation With Interleukin-1 Blockade in a Case of Erdheim-Chester Disease. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Guglielmi, B.; Berti, A.; Campochiaro, C.; Sabbadini, M.G.; Dagna, L. The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: Comprehensive review of the literature and of 10 new cases. Ann. Rheum. Dis. 2013, 72, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Wilejto, M.; Abla, O. Langerhans cell histiocytosis and Erdheim–Chester disease. Curr. Opin. Rheumatol. 2012, 24, 90–96. [Google Scholar] [CrossRef]
- Allen, C.E.; Merad, M.; McClain, K.L. Langerhans-Cell Histiocytosis. N. Engl. J. Med. 2018, 379, 856–868. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using Multiplexed Assays of Oncogenic Drivers in Lung Cancers to Select Targeted Drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Abdel-Wahab, O.; Durham, B.H. MAP-Kinase-Driven Hematopoietic Neoplasms: A Decade of Progress in the Molecular Age. Cold Spring Harb. Perspect. Med. 2020, a034892. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAFMutations in Hairy-Cell Leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Badalian-Very, G.; Vergilio, J.-A.; Degar, B.A.; MacConaill, L.E.; Brandner, B.; Calicchio, M.L.; Kuo, F.C.; Ligon, A.H.; Stevenson, K.E.; Kehoe, S.M.; et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010, 116, 1919–1923. [Google Scholar] [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.L.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Mazor, R.D.; Weissman, R.; Luckman, J.; Domachevsky, L.; Diamond, E.L.; Abdel-Wahab, O.; Shapira, S.; Hershkovitz-Rokah, O.; Groshar, D.; Shpilberg, O. Dual BRAF/MEK blockade restores CNS responses in BRAF-mutant Erdheim–Chester disease patients following BRAF inhibitor monotherapy. Neuro Oncol. Adv. 2020, 2. [Google Scholar] [CrossRef]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nat. Cell Biol. 2019, 567, 521–524. [Google Scholar] [CrossRef]
- Haroche, J.; Charlotte, F.; Arnaud, L.; Von Deimling, A.; Hélias-Rodzewicz, Z.; Hervier, B.; Aubart, F.C.; Launay, D.; Lesot, A.; Mokhtari, K.; et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012, 120, 2700–2703. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Durham, B.H.; Haroche, J.; Yao, Z.; Ma, J.; Parikh, S.A.; Wang, Z.; Choi, J.; Kim, E.; Cohen-Aubart, F.; et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016, 6, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Emile, J.-F.; Diamond, E.L.; Hélias-Rodzewicz, Z.; Cohen-Aubart, F.; Charlotte, F.; Hyman, D.M.; Kim, E.; Rampal, R.; Patel, M.; Ganzel, C.; et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood 2014, 124, 3016–3019. [Google Scholar] [CrossRef]
- Hyman, D.M.; Diamond, E.L.; Vibat, C.R.T.; Hassaine, L.; Poole, J.C.; Patel, M.; Holley, V.R.; Cabrilo, G.; Lu, T.T.; Arcila, M.E.; et al. Prospective Blinded Study of BRAF V600E Mutation Detection in Cell-Free DNA of Patients with Systemic Histiocytic Disorders. Cancer Discov. 2015, 5, 64–71. [Google Scholar] [CrossRef]
- Nordmann, T.M.; Juengling, F.D.; Recher, M.; Berger, C.T.; Kalbermatten, D.; Wicki, A.; Paasinen-Sohns, A.; Cathomas, G.; Tzankov, A.; Daikeler, T. Trametinib after disease reactivation under dabrafenib in Erdheim-Chester disease with both BRAF and KRAS mutations. Blood 2017, 129, 879–882. [Google Scholar] [CrossRef]
- Scheffzek, K. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef]
- Chakraborty, R.; Hampton, O.A.; Shen, X.; Simko, S.J.; Shih, A.; Abhyankar, H.; Lim, K.P.H.; Covington, K.R.; Trevino, L.; Dewal, N.; et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 2014, 124, 3007–3015. [Google Scholar] [CrossRef]
- Nelson, D.S.; Quispel, W.; Badalian-Very, G.; Van Halteren, A.G.; Bos, C.V.D.; Bovée, J.V.M.G.; Tian, S.Y.; Van Hummelen, P.; Ducar, M.; MacConaill, L.E.; et al. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood 2014, 123, 3152–3155. [Google Scholar] [CrossRef]
- Nelson, D.S.; Van Halteren, A.; Quispel, W.T.; Bos, C.V.D.; Bovée, J.V.M.G.; Patel, B.; Badalian-Very, G.; Van Hummelen, P.; Ducar, M.; Lin, L.; et al. MAP2K1 and MAP3K1 mutations in langerhans cell histiocytosis. Genes Chromosom. Cancer 2015, 54, 361–368. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nat. Cell Biol. 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Satoh, M.; Maesawa, C.; Takahashi, Y.; Tabuchi, T.; Itoh, T.; Nakamura, M. Effect of atorvastatin on microRNA 221/222 expression in endothelial progenitor cells obtained from patients with coronary artery disease. Eur. J. Clin. Investig. 2009, 39, 359–367. [Google Scholar] [CrossRef]
- Lu, C.; Huang, X.; Zhang, X.; Roensch, K.; Cao, Q.; Nakayama, K.I.; Blazar, B.R.; Zeng, Y.; Zhou, X. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood 2011, 117, 4293–4303. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Shukla, P.C.; Gupta, N.; Steer, B.M.; Ingram, A.J.; Gupta, M.; Al-Omran, M.; et al. MicroRNA-145 Targeted Therapy Reduces Atherosclerosis. Circulation 2012, 126, S81–S90. [Google Scholar] [CrossRef]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef]
- Mandolini, C.; Santovito, D.; Marcantonio, P.; Buttitta, F.; Bucci, M.; Ucchino, S.; Mezzetti, A.; Cipollone, F. Identification of microRNAs 758 and 33b as potential modulators of ABCA1 expression in human atherosclerotic plaques. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 202–209. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, Y.; Eom, S.; Kim, M.; Park, D.; Kim, H.; Noh, K.; Lee, H.; Lee, Y.S.; Choe, J.; et al. MicroRNA-26a/-26b-COX-2-MIP-2 Loop Regulates Allergic Inflammation and Allergic Inflammation-promoted Enhanced Tumorigenic and Metastatic Potential of Cancer Cells. J. Biol. Chem. 2015, 290, 14245–14266. [Google Scholar] [CrossRef] [PubMed]
- Raisch, J. Role of microRNAs in the immune system, inflammation and cancer. World J. Gastroenterol. 2013, 19, 2985–2996. [Google Scholar] [CrossRef]
- Mehta, A.; Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Poddar, S.; Kesharwani, D.; Datta, M. Interplay between the miRNome and the epigenetic machinery: Implications in health and disease. J. Cell. Physiol. 2017, 232, 2938–2945. [Google Scholar] [CrossRef]
- Berindan-Neagoe, I.; Bs, P.D.C.M.; Ms, B.P.; Calin, G.A. MicroRNAome genome: A treasure for cancer diagnosis and therapy. CA A Cancer J. Clin. 2014, 64, 311–336. [Google Scholar] [CrossRef]
- Drokow, E.K.; Sun, K.; Ahmed, H.A.W.; Akpabla, G.S.; Song, J.; Shi, M. Circulating microRNA as diagnostic biomarkers for haematological cancers: A systematic review and meta-analysis. Cancer Manag. Res. 2019, 11, 4313–4326. [Google Scholar] [CrossRef]
- Stanková, M.; Kubaczková, V.; Sedlarikova, L.; Sevcikova, S. Circulating microRNA as Biomarkers in Hematological Malignancies. Exp. Suppl. 2015, 106, 123–138. [Google Scholar] [CrossRef]
- Luna-Aguirre, C.M.; Martinez-Fierro, M.L.; Mar-Aguilar, F.; Garza-Veloz, I.; Treviño, V.; Rojas-Martinez, A.; Jaime-Pérez, J.C.; Malagon-Santiago, G.I.; Gutierrez-Aguirre, C.H.; Gonzalez-Llano, O.; et al. Circulating microRNA expression profile in B-cell acute lymphoblastic leukemia. Cancer Biomark. 2015, 15, 299–310. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Nonlinear partial differential equations and applications: Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Hao, D.; Liu, X.; Wang, D.; Ning, N.; Li, X. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nat. Cell Biol. 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Cheng, Y.Y.; Blenkiron, C.; Reid, G. Exploring Mechanisms of MicroRNA Downregulation in Cancer. MicroRNA 2017, 6, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef]
- Boyerinas, B.; Park, S.-M.; Hau, A.; Murmann, A.E.; Peter, M.E. The role of let-7 in cell differentiation and cancer. Endocr. Relat. Cancer 2010, 17, F19–F36. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Daley, G.Q. Lin28: A MicroRNA Regulator with a Macro Role. Cell 2010, 140, 445–449. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An Epigenetic Switch Involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 Links Inflammation to Cell Transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Mossetti, G.; Rendina, D.; Numis, F.G.; Somma, P.; Postiglione, L.; Nunziata, V. Biochemical markers of bone turnover, serum levels of interleukin-6/interleukin-6 soluble receptor and bisphosphonate treatment in Erdheim-Chester disease. Clin. Exp. Rheumatol. 2003, 21, 232–236. [Google Scholar]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS Is Regulated by the let-7 MicroRNA Family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Tsanov, K.M.; Pearson, D.S.; Wu, Z.; Han, A.; Triboulet, R.; Seligson, M.T.; Powers, J.T.; Osborne, J.K.; Kane, S.; Gygi, S.P.; et al. LIN28 phosphorylation by MAPK/ERK couples signalling to the post-transcriptional control of pluripotency. Nat. Cell Biol. 2017, 19, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Couts, K.L.; Anderson, E.M.; Gross, M.M.; Sullivan, K.; Ahn, N.G. Oncogenic B-Raf signaling in melanoma cells controls a network of microRNAs with combinatorial functions. Oncogene 2012, 32, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Perdas, E.; Stawski, R.; Kaczka, K.; Zubrzycka, M. Analysis of Let-7 Family miRNA in Plasma as Potential Predictive Biomarkers of Diagnosis for Papillary Thyroid Cancer. Diagnostics 2020, 10, 130. [Google Scholar] [CrossRef]
- Tang, H.; Ma, M.; Dai, J.; Cui, C.; Si, L.; Sheng, X.; Chi, Z.; Xu, L.; Yu, S.; Xu, T.; et al. miR-let-7b and miR-let-7c suppress tumourigenesis of human mucosal melanoma and enhance the sensitivity to chemotherapy. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.C.; Ricarte-Filho, J.C.M.; Fuziwara, C.S.; Yamashita, A.S.; Rezende, E.; Da-Silva, M.J.; Kimura, E.T. Effects of let-7 microRNA on Cell Growth and Differentiation of Papillary Thyroid Cancer. Transl. Oncol. 2009, 2, 236–241. [Google Scholar] [CrossRef]
- Kelly, B.D.; Miller, N.; Sweeney, K.J.; Durkan, G.C.; Rogers, E.; Walsh, K.; Kerin, M.J. A Circulating MicroRNA Signature as a Biomarker for Prostate Cancer in a High Risk Group. J. Clin. Med. 2015, 4, 1369–1379. [Google Scholar] [CrossRef]
- Tsujiura, M.; Ichikawa, D.; Komatsu, S.; Shiozaki, A.; Takeshita, H.; Kosuga, T.; Konishi, H.; Morimura, R.; Deguchi, K.; Fujiwara, H.; et al. Circulating microRNAs in plasma of patients with gastric cancers. Br. J. Cancer 2010, 102, 1174–1179. [Google Scholar] [CrossRef]
- Ghanbari, R.; Mosakhani, N.; Sarhadi, V.K.; Armengol, G.; Nouraee, N.; Mohammadkhani, A.; Khorrami, S.; Arefian, E.; Paryan, M.; Malekzadeh, R.; et al. Simultaneous Underexpression of let-7a-5p and let-7f-5p microRNAs in Plasma and Stool Samples from Early Stage Colorectal Carcinoma. Biomark. Cancer 2015, 7, 39–48. [Google Scholar] [CrossRef]
- Khalighfard, S.; Alizadeh, A.M.; Irani, S.; Omranipour, R. Plasma miR-21, miR-155, miR-10b, and Let-7a as the potential biomarkers for the monitoring of breast cancer patients. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Gianfreda, D.; Nicastro, M.; Galetti, M.; Alberici, F.; Corradi, D.; Becchi, G.; Baldari, G.; De Filippo, M.; Ferretti, S.; Moroni, G.; et al. Sirolimus plus prednisone for Erdheim-Chester disease: An open-label trial. Blood 2015, 126, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, F.; Maniscalco, V.; Peyronel, F.; Westenend, P.J.; Hendriksz, T.R.; Roperto, R.M.; Palumbo, A.A.; Sieni, E.; Romagnani, P.; Van Bommel, E.F.H.; et al. Long-term follow-up of mTOR inhibition for Erdheim-Chester disease. Blood 2020, 135, 1994–1997. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, D.; Shao, K.; Ye, B.; Huang, J.; Gao, Y. MiR-15a-5p negatively regulates cell survival and metastasis by targeting CXCL10 in chronic myeloid leukemia. Am. J. Transl. Res. 2017, 9, 4308–4316. [Google Scholar]
- Zeng, Y.-J.; Lai, W.; Wu, H.; Liu, L.; Xu, H.-Y.; Wang, J.; Chu, Z.-H. Neuroendocrine-like cells -derived CXCL10 and CXCL11 induce the infiltration of tumor-associated macrophage leading to the poor prognosis of colorectal cancer. Oncotarget 2016, 7, 27394–27407. [Google Scholar] [CrossRef]
- Lunardi, S.; Lim, S.Y.; Muschel, R.J.; Brunner, T.B. IP-10/CXCL10 attracts regulatory T cells: Implication for pancreatic cancer. OncoImmunology 2015, 4, e1027473. [Google Scholar] [CrossRef]
- Stoppacciaro, A.; Ferrarini, M.; Salmaggi, C.; Colarossi, C.; Praderio, L.; Tresoldi, M.; Beretta, A.A.; Sabbadini, M.G. Immunohistochemical evidence of a cytokine and chemokine network in three patients with Erdheim-Chester disease: Implications for pathogenesis. Arthritis Rheum. 2006, 54, 4018–4022. [Google Scholar] [CrossRef]
- Cavalli, G.; Biavasco, R.; Borgiani, B.; Dagna, L. Oncogene-Induced Senescence as a New Mechanism of Disease: The Paradigm of Erdheim-Chester Disease. Front. Immunol. 2014, 5, 281. [Google Scholar] [CrossRef]
- Shang, J.; He, Q.; Chen, Y.; Yu, D.; Sun, L.; Cheng, G.; Liu, D.; Xiao, J.; Zhao, Z. miR-15a-5p suppresses inflammation and fibrosis of peritoneal mesothelial cells induced by peritoneal dialysis via targeting VEGFA. J. Cell. Physiol. 2018, 234, 9746–9755. [Google Scholar] [CrossRef]
- Hu, H.Z.G.; Tian, G.; Lv, G.; Jin, Y. miRNA-15b-5p promotes expression of osteoblast differentiation-associated markers via targeting SMAD7. Int. J. Clin. Exp. Med. 2018, 11, 3578–3586. [Google Scholar]
- Vimalraj, S.; Partridge, N.C.; Selvamurugan, N. A Positive Role of MicroRNA-15b on Regulation of Osteoblast Differentiation. J. Cell. Physiol. 2014, 229, 1236–1244. [Google Scholar] [CrossRef]
- Yang, L.; Wang, B.; Zhou, Q.; Wang, Y.; Liu, X.; Liu, Z.; Zhan, Z. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Careccia, S.; Mainardi, S.; Pelosi, A.; Gurtner, A.; Diverio, D.; Riccioni, R.; Testa, U.; Pelosi, E.; Piaggio, G.; Sacchi, A.; et al. A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene 2009, 28, 4034–4040. [Google Scholar] [CrossRef]
- Pallasch, C.P.; Patz, M.; Park, Y.J.; Hagist, S.; Eggle, D.; Claus, R.; Debey-Pascher, S.; Schulz, A.; Frenzel, L.P.; Claasen, J.; et al. miRNA deregulation by epigenetic silencing disrupts suppression of the oncogene PLAG1 in chronic lymphocytic leukemia. Blood 2009, 114, 3255–3264. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Liu, X.; Xiong, X.; Zhang, C.; Li, J.; Zheng, H.; Huang, C.; Shi, Q.; Weng, Y. miR-107 promotes the erythroid differentiation of leukemia cells via the downregulation of Cacna2d1. Mol. Med. Rep. 2014, 11, 1334–1339. [Google Scholar] [CrossRef]
- Ji, Y.; Wei, Y.; Wang, J.; Ao, Q.; Gong, K.; Zuo, H. Decreased expression of microRNA-107 predicts poorer prognosis in glioma. Tumor Biol. 2015, 36, 4461–4466. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.-C.; Lin, S.-S.; Yuan, L.-J.; Lu, M.-L.; Ueng, S.W.N.; Yang, C.-Y.; Tsai, T.-T.; Lai, P.-L. Upregulation of miR-107 expression following hyperbaric oxygen treatment suppresses HMGB1/RAGE signaling in degenerated human nucleus pulposus cells. Arthritis Res. 2019, 21, 1–14. [Google Scholar] [CrossRef]
- Perez-Carbonell, L.; Sinicrope, F.A.; Alberts, S.R.; Oberg, A.L.; Balaguer, F.; Castells, A.; Boland, C.R.; Goel, A. MiR-320e is a novel prognostic biomarker in colorectal cancer. Br. J. Cancer 2015, 113, 83–90. [Google Scholar] [CrossRef]
- Chen, H.-F.; Chuang, H.-C.; Tan, T.-H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta Bioenergy 2007, 1773, 1227–1237. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2014, 43, D146–D152. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-J.; Chen, P.-J.; Duan, R.-Q.; Li, K.-J.; Wang, Y.-Z.; Li, Y. Up-regulation of miR-630 in clear cell renal cell carcinoma is associated with lower overall survival. Int. J. Clin. Exp. Pathol. 2014, 7, 3318–3323. [Google Scholar] [PubMed]
- Zhang, S.; Zhang, J.-Y.; Lu, L.-J.; Wang, C.-H.; Wang, L.-H. MiR-630 promotes epithelial ovarian cancer proliferation and invasion via targeting KLF6. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4542–4547. [Google Scholar]
- Durham, B.H.; Rodrigo, E.L.; Picarsic, J.; Abramson, D.; Rotemberg, V.; De Munck, S.; Pannecoucke, E.; Lu, S.X.; Pastore, A.; Yoshimi, A.; et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat. Med. 2019, 25, 1839–1842. [Google Scholar] [CrossRef]
- Pillar, N.; Polsky, A.L.; Weissglas-Volkov, D.; Shomron, N. Comparison of breast cancer metastasis models reveals a possible mechanism of tumor aggressiveness. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz-Rokah, O.; Modai, S.; Pasmanik-Chor, M.; Toren, A.; Shomron, N.; Raanani, P.; Shpilberg, O.; Granot, G. MiR-30e induces apoptosis and sensitizes K562 cells to imatinib treatment via regulation of the BCR–ABL protein. Cancer Lett. 2015, 356, 597–605. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Gender | Age (Years) | Involved Sites | Kinase Alteration |
|---|---|---|---|---|
| ECD1 | M | 75 | Bone, Peri-aortic Soft Tissue, Retroperitoneum | BRAF V600E |
| ECD2 | M | 35 | Bone, Brain, Cavernous sinus, Peri-aortic Soft Tissue, Retroperitoneum, Right Atrium, Skull Base | BRAF V600E |
| ECD3 | M | 50 | Bone, Brain, Peri-aortic Soft Tissue, Retroperitoneum, Right atrium | BRAF V600E |
| ECD4 | M | 75 | Bone, Orbit, Retroperitoneum | BRAF V600E |
| ECD5 | M | 58 | Bone, Orbit, Peri-aortic Soft Tissue, Retroperitoneum | BRAF V600E |
| ECD6 | M | 55 | Bone, Brain, Heart, Retroperitoneum, Testes | BRAF V600E |
| ECD7 | F | 56 | Bone, Brain, Retroperitoneum, Orbit | BRAF V600E |
| ECD8 | F | 83 | Bone, Dura, Orbit, Retroperitoneum, Skin | BRAF V600E |
| ECD9 | M | 53 | Bone, Peri-aortic, Retroperitoneum | BRAF V600E |
| ECD10 | F | 52 | Bone, Brain | BRAF V600E |
| ECD11 | M | 38 | Bone, Brain, Dura, Heart, Peri-aortic, Retroperitoneum, Skin, Skull base | BRAF V600E |
| ECD12 | M | 46 | Bone, Brain, Retroperitoneum | BRAF V600E |
| ECD13 | M | 67 | Bone, Dura, Lymph Nodes, Peri-aortic, Retroperitoneum | NRAS Q61R |
| ECD14 | F | 66 | Bone, Heart | KRAS G12S |
| ECD15 | M | 69 | Bone, Retroperitoneum, Subcutaneous Soft Tissues | MAP2K2 Y134H |
| ECD16 | M | 69 | Bone, Brain, Peri-aortic, Retroperitoneum | MAP2K1 C121S |
| ECD17 | M | 18 | Bone, Brain, Dura | BRAF V600E |
| ECD18 | F | 77 | Bone, Brain | BRAF V600E |
| ECD19 | M | 51 | Bone, Larynx, Subcutaneous Soft Tissues | KRAS G12R/ARAF P216A |
| ECD20 | M | 57 | Bone, Pituitary, Retroperitoneum, Spleen | MAP2K1 Q56P |
| ECD21 | M | 54 | Bone, Brain, Heart, Retroperitoneum | BRAF V600E |
| ECD22 | M | 47 | Bone, Brain, Heart, Pleura, Retroperitoneum, Spine | KRAS R149G |
| ECD23 | F | 59 | Bone, Heart, Lymph Nodes, Pleura, Retroperitoneum, Skin | Unknown |
| ECD24 | F | 66 | Bone, Heart, Peri-aortic | MAP2K1 P124Q |
| ECD25 | F | 35 | Bone, Brain | BRAF N486_P490del |
| ECD26 | M | 48 | Bone, Mucosa | Unknown |
| ECD27 | F | 45 | Adrenal, Bone, Heart | BRAF V600E |
| ECD28 | M | 58 | Brain, Sinus, Skin | BRAF V600E |
| ECD29 | M | 63 | Kidney, Skin | Unknown |
| ECD30 | M | 76 | Bone, Brain, Sinus | BRAF V600E |
| ECD31 | F | 39 | Heart | BRAF V600E |
| ECD32 * | F | 76 | Brain, Heart, Kidney, Lungs | BRAF V600E |
| LCH1 | F | 40 | Bone, Lung | BRAF V600E |
| LCH2 | F | 72 | Bone, Brain, Skin, Vulva | BRAF V600E |
| LCH3 | F | 34 | Bone | BRAF V600E |
| LCH4 | F | 34 | Bone, Lymph Nodes, Skin, Subcutaneous Soft Tissues | BRAF V600E |
| LCH5 | M | 57 | Colon, Oral Mucosa, Skin | BRAF N486_P490del |
| LCH6 | M | 58 | Bone, Lymph Node, Mastoid, Pancreas, Spine, Submandibular gland | BICD2-BRAF fusion |
| LCH7 | M | 33 | Bone, Lung, Skin | BRAF V600E |
| Name | Gender | Age (Years) | Involved Sites | Kinase Alteration | Biopsy Site |
|---|---|---|---|---|---|
| ECD15 | M | 69 | Bone, retroperitoneum, subcutaneous soft tissues | MAP2K2 Y134H | Tibia |
| ECD21 | M | 54 | Bone, brain, heart, retroperitoneum | BRAF V600E | Cerebellum |
| ECD33 * | M | 34 | Bone, brain, gallbladder and bile ducts, Kidney, lungs, omentum, mesenterium retroperitoneum | BRAF V600E | Omentum |
| ECD34 | M | 75 | Kidney, pancreas, peri-aortic, retroperitoneum | Unknown | Peri-nepheric |
| ECD35 | M | 67 | Aorta, kidney | MAP2K1 C121S | Peri-nepheric |
| LCH8 * | M | 34 | Bone, brain, gallbladder and bile ducts, kidney, lungs, omentum retroperitoneum | BRAF V600E | Gall bladder |
| LCH9 | F | 53 | Rib | BRAF V600E | Rib |
| LCH10 | F | 24 | Gums, lungs | BRAF V600E+ FAT1 | Oral Mucosa |
| LCH11 | F | 30 | Bone, mucosa, scalp | MAP2K1 E102_I103 del | Gingiva |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weissman, R.; Diamond, E.L.; Haroche, J.; Pillar, N.; Shapira, G.; Durham, B.H.; Buthorn, J.; Cohen, F.; Ki, M.; Stemer, G.; et al. The Contribution of MicroRNAs to the Inflammatory and Neoplastic Characteristics of Erdheim–Chester Disease. Cancers 2020, 12, 3240. https://doi.org/10.3390/cancers12113240
Weissman R, Diamond EL, Haroche J, Pillar N, Shapira G, Durham BH, Buthorn J, Cohen F, Ki M, Stemer G, et al. The Contribution of MicroRNAs to the Inflammatory and Neoplastic Characteristics of Erdheim–Chester Disease. Cancers. 2020; 12(11):3240. https://doi.org/10.3390/cancers12113240
Chicago/Turabian StyleWeissman, Ran, Eli L. Diamond, Julien Haroche, Nir Pillar, Guy Shapira, Benjamin H. Durham, Justin Buthorn, Fleur Cohen, Michelle Ki, Galia Stemer, and et al. 2020. "The Contribution of MicroRNAs to the Inflammatory and Neoplastic Characteristics of Erdheim–Chester Disease" Cancers 12, no. 11: 3240. https://doi.org/10.3390/cancers12113240
APA StyleWeissman, R., Diamond, E. L., Haroche, J., Pillar, N., Shapira, G., Durham, B. H., Buthorn, J., Cohen, F., Ki, M., Stemer, G., Ulaner, G. A., Amoura, Z., Emile, J.-F., Mazor, R. D., Shomron, N., Abdel-Wahab, O. I., Shpilberg, O., & Hershkovitz-Rokah, O. (2020). The Contribution of MicroRNAs to the Inflammatory and Neoplastic Characteristics of Erdheim–Chester Disease. Cancers, 12(11), 3240. https://doi.org/10.3390/cancers12113240