LncRNAs in Cancer: From garbage to Junk





{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
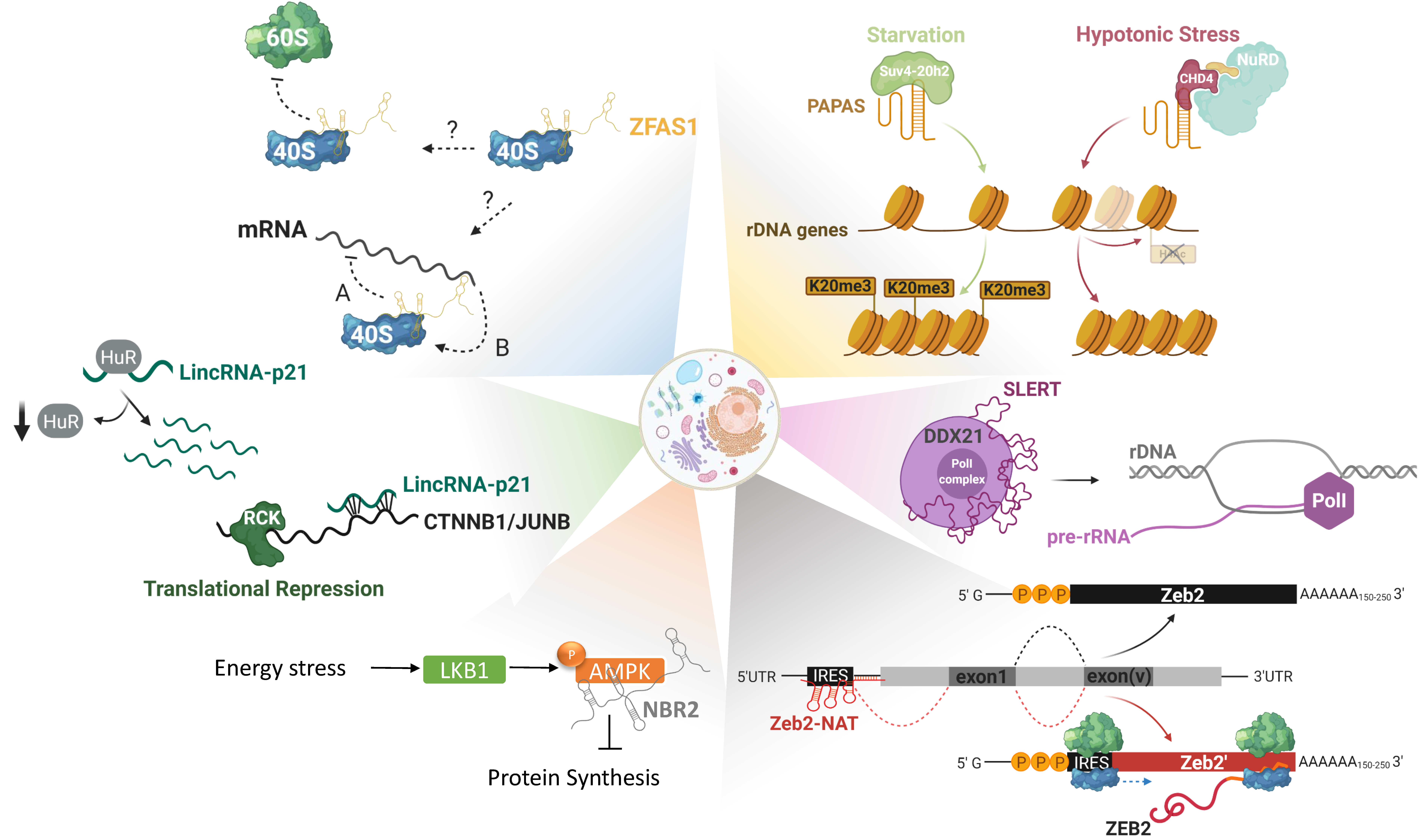
2. Epigenetic Regulation and Translational Reprogramming
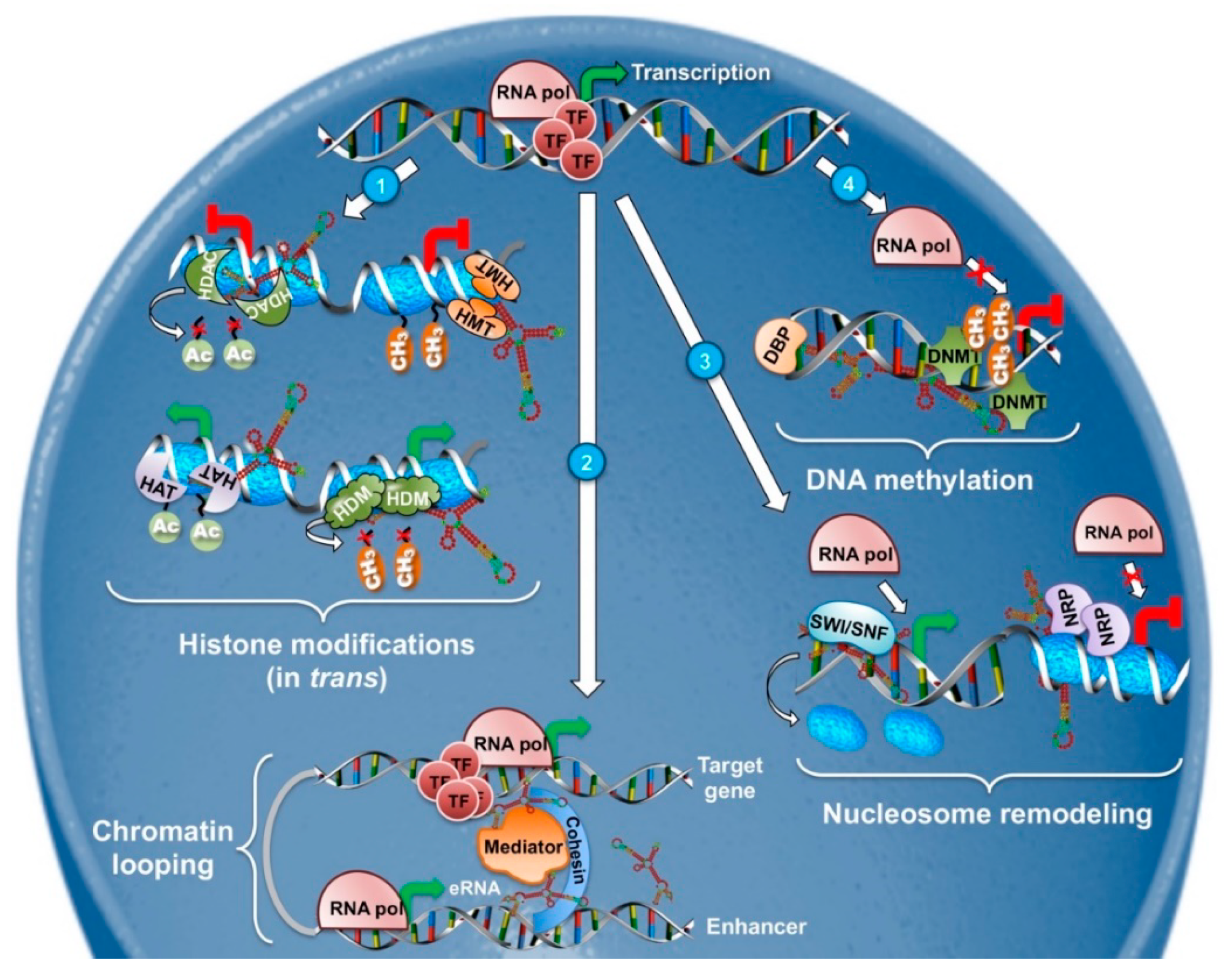
2.1. LncRNAs in Epigenetic and Transcriptional Regulation (DNA Methylation, Nucleosome Positioning, and Histone Modifications)
2.1.1. DNA Methylation
2.1.2. Histone Modifications
2.1.3. Chromatin Looping
2.1.4. Nucleosome Positioning
2.2. LncRNA Acting as ceRNAs
2.3. LncRNAs in Translational Reprogramming
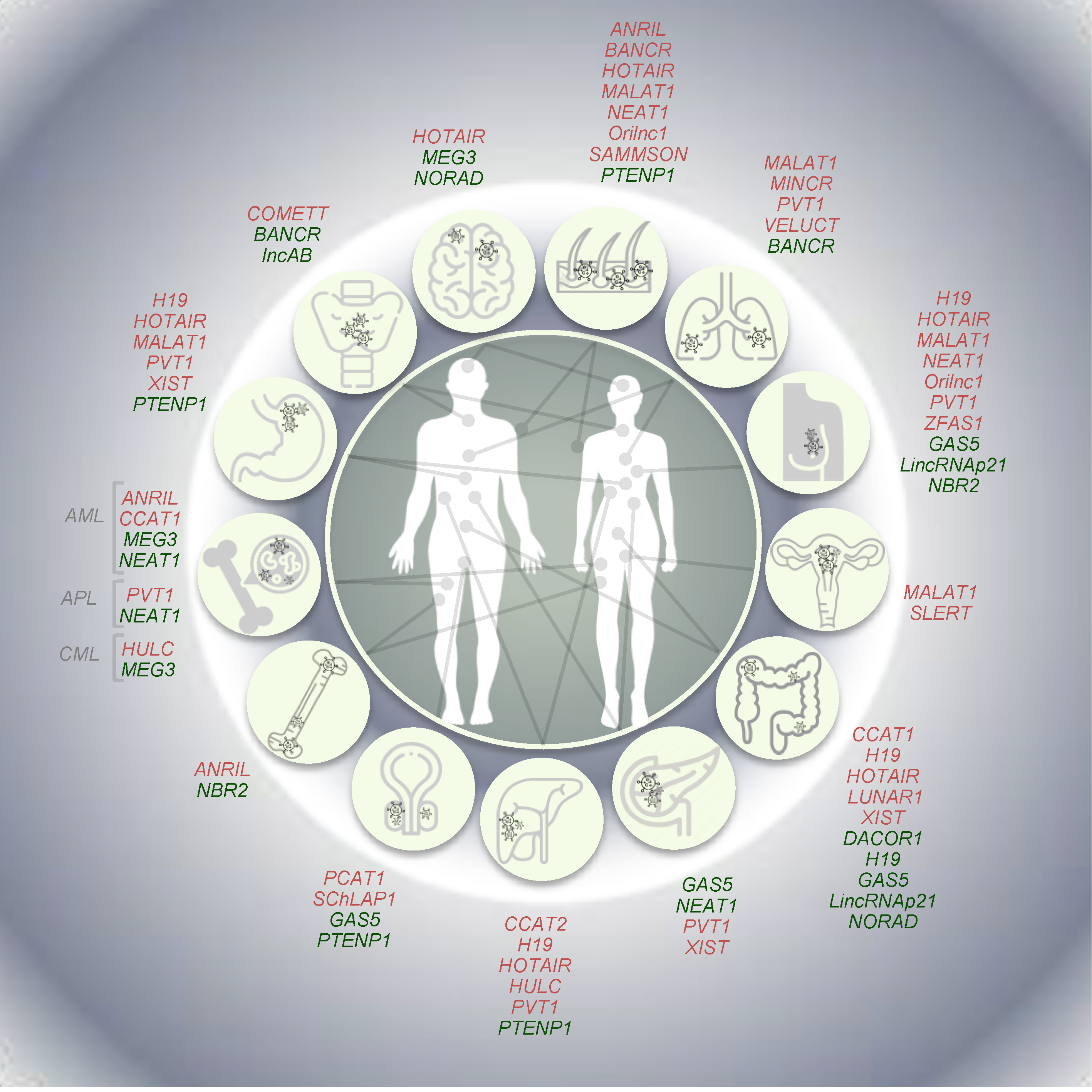
3. LncRNAs in Tumorigenesis
3.1. Oncogenic LncRNAs
3.2. Tumor Suppressive lncRNAs
3.3. LncRNAs with Dual Activity
4. Strategies to Target lncRNAs
4.1. Genomic Modulation of LncRNAs by CRISPR-Based Systems
4.2. Post-Transcriptional Targeting of LncRNAs
4.3. Steric Inhibition of LncRNA-Protein Interactions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Euskirchen, G.; Bertone, P.; Martone, R.; Luscombe, N.M.; Hartman, S.; Harrison, P.M.; Nelson, F.K.; Miller, P.; Gerstein, M.; et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003, 17, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 457, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [PubMed]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Moran, V.A.; Perera, R.J.; Khalil, A.M. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012, 40, 6391–6400. [Google Scholar] [CrossRef]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef]
- Khurana, E.; Fu, Y.; Colonna, V.; Mu, X.J.; Kang, H.M.; Lappalainen, T.; Sboner, A.; Lochovsky, L.; Chen, J.; Harmanci, A.; et al. Integrative annotation of variants from 1092 humans: Application to cancer genomics. Science 2013, 342, 1235587. [Google Scholar] [CrossRef]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, X.; Zhi, H.; Zhang, Y.; Wang, P.; Wang, Y.; Shang, S.; Fang, Y.; Shen, W.; Ning, S.; et al. Comprehensive Characterization of Somatic Mutations Impacting lncRNA Expression for Pan-Cancer. Mol. Nucleic. Acids 2019, 18, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef]
- Niland, C.N.; Merry, C.R.; Khalil, A.M. Emerging Roles for Long Non-Coding RNAs in Cancer and Neurological Disorders. Front. Genet. 2012, 3, 25. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Adriaens, C.; Standaert, L.; Barra, J.; Latil, M.; Verfaillie, A.; Kalev, P.; Boeckx, B.; Wijnhoven, P.W.G.; Radaelli, E.; Vermi, W.; et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 2016, 22, 861–868. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Mattick, J.S. The central role of RNA in human development and cognition. Febs Lett. 2011, 585, 1600–1616. [Google Scholar] [CrossRef]
- Derrien, T. The long non-coding RNAs: A new (p)layer in the “dark matter”. Front. Genet. 2012, 2, 107. [Google Scholar] [CrossRef]
- Wilson, K.D.; Ameen, M.; Guo, H.; Abilez, O.J.; Tian, L.; Mumbach, M.R.; Diecke, S.; Qin, X.; Liu, Y.; Yang, H.; et al. Endogenous Retrovirus-Derived lncRNA BANCR Promotes Cardiomyocyte Migration in Humans and Non-human Primates. Dev. Cell 2020, 54, 694–709.e9. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.-V.; Brown, E.L.; DeSouza, T.; Jespersen, N.Z.; Nandrup-Bus, C.; Yang, Q.; Yang, Z.; Desai, A.; Min, S.Y.; Rojas-Rodriguez, R.; et al. Human thermogenic adipocyte regulation by the long noncoding RNA LINC00473. Nat. Metab. 2020, 2, 397–412. [Google Scholar] [CrossRef]
- Nakagawa, S.; Naganuma, T.; Shioi, G.; Hirose, T. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 2011, 193, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Horvat, F.; Drutovic, D.; Efenberkova, M.; Pinkas, D.; Jindrova, A.; Pasulka, J.; Iyyappan, R.; Malik, R.; Susor, A.; et al. The most abundant maternal lncRNA Sirena1 acts post-transcriptionally and impacts mitochondrial distribution. Nucleic Acids Res. 2020, 48, 3211–3227. [Google Scholar] [CrossRef]
- Verheyden, Y.; Goedert, L.; Leucci, E. Control of nucleolar stress and translational reprogramming by lncRNAs. Cell Stress 2018, 3, 19–26. [Google Scholar] [CrossRef]
- Barra, J.; Gaidosh, G.S.; Blumenthal, E.; Beckedorff, F.; Tayari, M.M.; Kirstein, N.; Karakach, T.K.; Jensen, T.H.; Impens, F.; Gevaert, K.; et al. Integrator restrains paraspeckles assembly by promoting isoform switching of the lncRNA NEAT1. Sci. Adv. 2020, 6, eaaz9072. [Google Scholar] [CrossRef]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.S.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef]
- Van Heesch, S.; van Iterson, M.; Jacobi, J.; Boymans, S.; Essers, P.B.; de Bruijn, E.; Hao, W.; MacInnes, A.W.; Cuppen, E.; Simonis, M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014, 15, R6. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Shah, A.; Shan, G. Long Non-coding RNAs in the Cytoplasm. Genom. Proteom. Bioinform. 2016, 14, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Fraser, P. No-nonsense functions for long noncoding RNAs. Cell 2011, 145, 178–181. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Begolli, R.; Sideris, N.; Giakountis, A. LncRNAs as Chromatin Regulators in Cancer: From Molecular Function to Clinical Potential. Cancers 2019, 11, 1524. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; De Figueiredo Pontes, L.L.; Alberich-Jorda, M.; Zhang, P.; et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef]
- Merry, C.R.; Forrest, M.E.; Sabers, J.N.; Beard, L.; Gao, X.-H.; Hatzoglou, M.; Jackson, M.W.; Wang, Z.; Markowitz, S.D.; Khalil, A.M. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 2015, 24, 6240–6253. [Google Scholar] [CrossRef]
- Somasundaram, S.; Forrest, M.E.; Moinova, H.; Cohen, A.; Varadan, V.; LaFramboise, T.; Markowitz, S.; Khalil, A.M. The DNMT1-associated lincRNA DACOR1 reprograms genome-wide DNA methylation in colon cancer. Clin. Epigenetics 2018, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhi, H.; Li, X.; Wang, P.; Gao, Y.; Gao, B.; Zhou, D.; Zhang, Y.; Guo, M.; Yue, M.; Shen, W.; et al. Lnc2Meth: A manually curated database of regulatory relationships between long non-coding RNAs and DNA methylation associated with human disease. Nucleic Acids Res. 2018, 46, D133–D138. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, S. The nucleosome: From genomic organization to genomic regulation. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef]
- Ozdağ, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Nat. Publ. Group 2011, 30, 3391–3403. [Google Scholar] [CrossRef]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Wagschal, A.; Sutherland, H.G.; Woodfine, K.; Henckel, A.; Chebli, K.; Schulz, R.; Oakey, R.J.; Bickmore, W.A.; Feil, R. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol. Cell. Biol. 2008, 28, 1104–1113. [Google Scholar] [CrossRef]
- Fan, J.; Xing, Y.; Wen, X.; Jia, R.; Ni, H.; He, J.; Ding, X.; Pan, H.; Qian, G.; Ge, S.; et al. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 2015, 16, 1–17. [Google Scholar]
- Borsani, G.; Tonlorenzi, R.; Simmler, M.C.; Dandolo, L.; Arnaud, D.; Capra, V.; Grompe, M.; Pizzuti, A.; Muzny, D.; Lawrence, C.; et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature 1991, 351, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafrenière, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Payer, B.; Lee, J.T. X chromosome dosage compensation: How mammals keep the balance. Annu. Rev. Genet. 2008, 42, 733–772. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A. Gene silencing in X-chromosome inactivation: Advances in understanding facultative heterochromatin formation. Nat. Rev. Genet. 2011, 12, 542–553. [Google Scholar] [CrossRef]
- Barr, M.L.; Bertram, E.G. A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 1949, 163, 676–677. [Google Scholar] [CrossRef]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef]
- Liao, D.J.; Du, Q.-Q.; Yu, B.W.; Grignon, D.; Sarkar, F.H. Novel perspective: Focusing on the X chromosome in reproductive cancers. Cancer Investig. 2003, 21, 641–658. [Google Scholar] [CrossRef]
- Pageau, G.J.; Hall, L.L.; Ganesan, S.; Livingston, D.M.; Lawrence, J.B. The disappearing Barr body in breast and ovarian cancers. Nat. Rev. Cancer 2007, 7, 628–633. [Google Scholar] [CrossRef]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Nat. Publ. Group 2011, 30, 1956–1962. [Google Scholar]
- Shete, S.; Hosking, F.J.; Robertson, L.B.; Dobbins, S.E.; Sanson, M.; Malmer, B.; Simon, M.; Marie, Y.; Boisselier, B.; Delattre, J.-Y.; et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 2009, 41, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Stacey, S.N.; Sulem, P.; Masson, G.; Gudjonsson, S.A.; Thorleifsson, G.; Jakobsdottir, M.; Sigurdsson, A.; Gudbjartsson, D.F.; Sigurgeirsson, B.; Benediktsdottir, K.R.; et al. New common variants affecting susceptibility to basal cell carcinoma. Nat. Genet. 2009, 41, 909–914. [Google Scholar] [CrossRef]
- Turnbull, C.; Ahmed, S.; Morrison, J.; Pernet, D.; Renwick, A.; Maranian, M.; Seal, S.; Ghoussaini, M.; Hines, S.; Healey, C.S.; et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010, 42, 504–507. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Vidaud, M.; Bièche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. Faseb J. 2011, 25, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Davidovich, C.; Zheng, L.; Goodrich, K.J.; Cech, T.R. Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 2013, 20, 1250–1257. [Google Scholar] [CrossRef]
- Kretz, M.; Meister, G. RNA binding of PRC2: Promiscuous or well ordered? Mol. Cell 2014, 55, 157–158. [Google Scholar] [CrossRef]
- Davidovich, C.; Wang, X.; Cifuentes-Rojas, C.; Goodrich, K.J.; Gooding, A.R.; Lee, J.T.; Cech, T.R. Toward a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol. Cell 2015, 57, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Portoso, M.; Ragazzini, R.; Brenčič, Ž.; Moiani, A.; Michaud, A.; Vassilev, I.; Wassef, M.; Servant, N.; Sargueil, B.; Margueron, R. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. Embo J. 2017, 36, 981–994. [Google Scholar] [CrossRef]
- Jain, A.K.; Xi, Y.; McCarthy, R.; Allton, K.; Akdemir, K.C.; Patel, L.R.; Aronow, B.; Lin, C.; Li, W.; Yang, L.; et al. LncPRESS1 Is a p53-Regulated LncRNA that Safeguards Pluripotency by Disrupting SIRT6-Mediated De-acetylation of Histone H3K56. Mol. Cell 2016, 64, 967–981. [Google Scholar] [CrossRef]
- Plank, J.L.; Dean, A. Enhancer function: Mechanistic and genome-wide insights come together. Mol. Cell 2014, 55, 5–14. [Google Scholar] [CrossRef]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Barozzi, I.; Mietton, F.; Ghisletti, S.; Polletti, S.; Tusi, B.K.; Muller, H.; Ragoussis, J.; Wei, C.-L.; Natoli, G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010, 8, e1000384. [Google Scholar] [CrossRef]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef]
- Hah, N.; Danko, C.G.; Core, L.; Waterfall, J.J.; Siepel, A.; Lis, J.T.; Kraus, W.L. A Rapid, Extensive, and Transient Transcriptional Response to Estrogen Signaling in Breast Cancer Cells. Cell 2011, 145, 622–634. [Google Scholar] [CrossRef]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 2016, 15, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Leong, W.Z.; Ngoc, P.C.T.; Tan, T.K.; Bertulfo, F.C.; Lim, M.C.; An, O.; Li, Z.; Yeoh, A.E.J.; Fullwood, M.J.; et al. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 2019, 134, 239–251. [Google Scholar] [CrossRef]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef]
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.-C.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J. Exp. Med. 2011, 208, 1809–1822. [Google Scholar] [CrossRef]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of Chromatin Structure by Long Noncoding RNA. Trends Cell Biol. 2015, 25, 623–632. [Google Scholar] [CrossRef]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef]
- Vydzhak, O.; Luke, B.; Schindler, N. Non-coding RNAs at the Eukaryotic rDNA Locus: RNA-DNA Hybrids and Beyond. J. Mol. Biol. 2020, 432, 4287–4304. [Google Scholar] [CrossRef]
- Bierhoff, H.; Dammert, M.A.; Brocks, D.; Dambacher, S.; Schotta, G.; Grummt, I. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol. Cell 2014, 54, 675–682. [Google Scholar] [CrossRef]
- Zhao, Z.; Dammert, M.A.; Grummt, I.; Bierhoff, H. lncRNA-Induced Nucleosome Repositioning Reinforces Transcriptional Repression of rRNA Genes upon Hypotonic Stress. Cell Rep. 2016, 14, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sentürk, N.; Song, C.; Grummt, I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018, 32, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In Vivo Identification of Tumor- Suppressive PTEN ceRNAs in an Oncogenic BRAF-Induced Mouse Model of Melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Sumazin, P.; Yang, X.; Chiu, H.-S.; Chung, W.-J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An Extensive MicroRNA-Mediated Network of RNA-RNA Interactions Regulates Established Oncogenic Pathways in Glioblastoma. Cell 2011, 147, 370–381. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, J.; He, D.; Sun, M.; Zhang, P.; Yu, Y.; Chen, Y. The Emerging Function and Mechanism of ceRNAs in Cancer. Trends Genet. 2016, 32, 211–224. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Li, R.-K.; Gao, J.; Guo, L.-H.; Huang, G.-Q.; Luo, W.-H. PTENP1 acts as a ceRNA to regulate PTEN by sponging miR-19b and explores the biological role of PTENP1 in breast cancer. Cancer Gene 2017, 24, 309–315. [Google Scholar] [CrossRef]
- Gao, L.; Ren, W.; Zhang, L.; Li, S.; Kong, X.; Zhang, H.; Dong, J.; Cai, G.; Jin, C.; Zheng, D.; et al. PTENp1, a natural sponge of miR-21, mediates PTEN expression to inhibit the proliferation of oral squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1322–1334. [Google Scholar] [CrossRef]
- Poliseno, L.; Haimovic, A.; Christos, P.J.; Vega Y Saenz de Miera, E.C.; Shapiro, R.; Pavlick, A.; Berman, R.S.; Darvishian, F.; Osman, I. Deletion of PTENP1 pseudogene in human melanoma. J. Investig. Derm. 2011, 131, 2497–2500. [Google Scholar] [CrossRef]
- Panzitt, K.; Tschernatsch, M.M.O.; Guelly, C.; Moustafa, T.; Stradner, M.; Strohmaier, H.M.; Buck, C.R.; Denk, H.; Schroeder, R.; Trauner, M.; et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 2007, 132, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef]
- Lu, Z.; Xiao, Z.; Liu, F.; Cui, M.; Li, W.; Yang, Z.; Li, J.; Ye, L.; Zhang, X. Long non-coding RNA HULC promotes tumor angiogenesis in liver cancer by up-regulating sphingosine kinase 1 (SPHK1). Oncotarget 2016, 7, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Y.; Chai, X.; Kang, Q.; Zhao, P.; Xiong, J.; Wang, J. Long noncoding RNA HULC promotes cell proliferation by regulating PI3K/AKT signaling pathway in chronic myeloid leukemia. Gene 2017, 607, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zheng, H.; Chan, M.T.V.; Wu, W.K.K. HULC: An oncogenic long non-coding RNA in human cancer. J. Cell Mol. Med. 2017, 21, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef] [PubMed]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect Biol. 2019, 11, a032896. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef]
- Shaulian, E. AP-1--The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Fu, Y.; Zheng, S.; An, N.; Athanasopoulos, T.; Popplewell, L.; Liang, A.; Li, K.; Hu, C.; Zhu, Y. β-catenin as a potential key target for tumor suppression. Int. J. Cancer 2011, 129, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lei, Z.-J.; Guo, Y.; Wang, T.; Qin, Z.-Y.; Xiao, H.-L.; Fan, L.-L.; Chen, D.-F.; Bian, X.-W.; Liu, J.; et al. miRNA-regulated delivery of lincRNA-p21 suppresses β-catenin signaling and tumorigenicity of colorectal cancer stem cells. Oncotarget 2015, 6, 37852–37870. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Yang, X.; Li, J.; Yang, W.; Ma, H.; Zhang, Z. p53-targeted lincRNA-p21 acts as a tumor suppressor by inhibiting JAK2/STAT3 signaling pathways in head and neck squamous cell carcinoma. Mol. Cancer 2019, 18, 38. [Google Scholar] [CrossRef]
- Xing, Y.-H.; Yao, R.-W.; Zhang, Y.; Guo, C.-J.; Jiang, S.; Xu, G.; Dong, R.; Yang, L.; Chen, L.-L. SLERT Regulates DDX21 Rings Associated with Pol I Transcription. Cell 2017, 169, 664–678.e16. [Google Scholar] [CrossRef]
- Hansji, H.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Cameron-Smith, D.; Figueiredo, V.C.; Askarian-Amiri, M.E. ZFAS1: A long noncoding RNA associated with ribosomes in breast cancer cells. Biol. Direct 2016, 11, 62. [Google Scholar] [CrossRef]
- Liu, B.; Qian, S.-B. Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–315. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Leppek, K.; Das, R.; Barna, M. Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Alvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef]
- Lan, T.; Chang, L.; Wu, L.; Yuan, Y. Downregulation of ZEB2-AS1 decreased tumor growth and metastasis in hepatocellular carcinoma. Mol. Med. Rep. 2016, 14, 4606–4612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, H.; Sun, R.; Li, P.; Yang, Z.; Liu, Y.; Wang, Z.; Yang, Y.; Yin, C. Long non-coding RNA ZEB2-AS1 promotes the proliferation, metastasis and epithelial mesenchymal transition in triple-negative breast cancer by epigenetically activating ZEB2. J. Cell Mol. Med. 2019, 23, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, Z.-D.; Han, L.; Zhang, J.; Lee, S.-W.; Wang, W.; Lee, H.; Zhuang, L.; Chen, J.; Lin, H.-K.; et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat. Cell Biol. 2016, 18, 431–442. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef]
- Carlevaro-Fita, J.; Lanzós, A.; Feuerbach, L.; Hong, C.; Mas-Ponte, D.; Pedersen, J.S.; PCAWG Drivers and Functional Interpretation Group; Johnson, R.; PCAWG Consortium. Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis. Commun. Biol. 2020, 3, 56. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, L.; Wu, L.-M.; Lai, M.-C.; Xie, H.-Y.; Zhang, F.; Zheng, S.-S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef]
- Gutschner, T.; Hämmerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef]
- Okugawa, Y.; Toiyama, Y.; Hur, K.; Toden, S.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Mohri, Y.; Kusunoki, M.; Boland, C.R.; et al. Metastasis-associated long non-coding RNA drives gastric cancer development and promotes peritoneal metastasis. Carcinogenesis 2014, 35, 2731–2739. [Google Scholar] [CrossRef]
- Yang, L.; Bai, H.-S.; Deng, Y.; Fan, L. High MALAT1 expression predicts a poor prognosis of cervical cancer and promotes cancer cell growth and invasion. Eur. Rev. Med. Pharm. Sci. 2015, 19, 3187–3193. [Google Scholar]
- Li, C.; Cui, Y.; Liu, L.-F.; Ren, W.-B.; Li, Q.-Q.; Zhou, X.; Li, Y.-L.; Li, Y.; Bai, X.-Y.; Zu, X.-B. High Expression of Long Noncoding RNA MALAT1 Indicates a Poor Prognosis and Promotes Clinical Progression and Metastasis in Bladder Cancer. Clin. Genitourin. Cancer 2017, 15, 570–576. [Google Scholar] [CrossRef]
- Jin, Y.; Feng, S.-J.; Qiu, S.; Shao, N.; Zheng, J.-H. LncRNA MALAT1 promotes proliferation and metastasis in epithelial ovarian cancer via the PI3K-AKT pathway. Eur. Rev. Med. Pharm. Sci. 2017, 21, 3176–3184. [Google Scholar]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Jadaliha, M.; Zong, X.; Malakar, P.; Ray, T.; Singh, D.K.; Freier, S.M.; Jensen, T.; Prasanth, S.G.; Karni, R.; Ray, P.S.; et al. Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget 2016, 7, 40418–40436. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Schorderet, P.; Duboule, D. Structural and functional differences in the long non-coding RNA hotair in mouse and human. PLoS Genet. 2011, 7, e1002071. [Google Scholar] [CrossRef]
- Somarowthu, S.; Legiewicz, M.; Chillón, I.; Marcia, M.; Liu, F.; Pyle, A.M. HOTAIR forms an intricate and modular secondary structure. Mol. Cell 2015, 58, 353–361. [Google Scholar] [CrossRef]
- Sharma, S.; Mandal, P.; Sadhukhan, T.; Roy Chowdhury, R.; Ranjan Mondal, N.; Chakravarty, B.; Chatterjee, T.; Roy, S.; Sengupta, S. Bridging Links between Long Noncoding RNA HOTAIR and HPV Oncoprotein E7 in Cervical Cancer Pathogenesis. Sci. Rep. 2015, 5, 11724. [Google Scholar] [CrossRef]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, R.; Verheyden, Y.; Ishikawa, H.; Goedert, L.; Nicolas, E.; Saraf, K.; Armaos, A.; Delli Ponti, R.; Izumikawa, K.; Mestdagh, P.; et al. SAMMSON fosters cancer cell fitness by concertedly enhancing mitochondrial and cytosolic translation. Nat. Struct. Mol. Biol. 2018, 25, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.; Breinig, M.; Caudron-Herger, M.; Polycarpou-Schwarz, M.; Boutros, M.; Diederichs, S. The lncRNA VELUCT strongly regulates viability of lung cancer cells despite its extremely low abundance. Nucleic Acids Res. 2017, 45, 5458–5469. [Google Scholar] [CrossRef]
- Shtivelman, E.; Henglein, B.; Groitl, P.; Lipp, M.; Bishop, J.M. Identification of a human transcription unit affected by the variant chromosomal translocations 2;8 and 8;22 of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1989, 86, 3257–3260. [Google Scholar] [CrossRef]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafà, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef]
- Kim, T.; Cui, R.; Jeon, Y.-J.; Lee, J.-H.; Lee, J.H.; Sim, H.; Park, J.K.; Fadda, P.; Tili, E.; Nakanishi, H.; et al. Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc. Natl. Acad. Sci. USA 2014, 111, 4173–4178. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chen, W.; Han, S.; Iyer, M.K.; Cao, Q.; Kothari, V.; Evans, J.R.; Knudsen, K.E.; Paulsen, M.T.; Ljungman, M.; et al. The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia 2014, 16, 900–908. [Google Scholar] [CrossRef]
- Doose, G.; Haake, A.; Bernhart, S.H.; López, C.; Duggimpudi, S.; Wojciech, F.; Bergmann, A.K.; Borkhardt, A.; Burkhardt, B.; Claviez, A.; et al. MINCR is a MYC-induced lncRNA able to modulate MYC’s transcriptional network in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5261–E5270. [Google Scholar] [CrossRef]
- Huppi, K.; Pitt, J.J.; Wahlberg, B.M.; Caplen, N.J. The 8q24 gene desert: An oasis of non-coding transcriptional activity. Front. Genet. 2012, 3, 69. [Google Scholar] [CrossRef]
- Iaccarino, I. lncRNAs and MYC: An Intricate Relationship. Int. J. Mol. Sci. 2017, 18, 1497. [Google Scholar] [CrossRef]
- Webb, E.; Adams, J.M.; Cory, S. Variant (6;15) translocation in a murine plasmacytoma occurs near an immunoglobulin kappa gene but far from the myc oncogene. Nature 1984, 312, 777–779. [Google Scholar] [CrossRef]
- Cory, S.; Graham, M.; Webb, E.; Corcoran, L.; Adams, J.M. Variant (6;15) translocations in murine plasmacytomas involve a chromosome 15 locus at least 72 kb from the c-myc oncogene. Embo J. 1985, 4, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Bishop, J.M. The PVT gene frequently amplifies with MYC in tumor cells. Mol. Cell. Biol. 1989, 9, 1148–1154. [Google Scholar] [CrossRef]
- Graham, M.; Adams, J.M. Chromosome 8 breakpoint far 3” of the c-myc oncogene in a Burkitt”s lymphoma 2;8 variant translocation is equivalent to the murine pvt-1 locus. Embo J. 1986, 5, 2845–2851. [Google Scholar] [CrossRef]
- Siwarski, D.; Müller, U.; Andersson, J.; Notario, V.; Melchers, F.; Rolink, A.; Huppi, K. Structure and expression of the c-Myc/Pvt 1 megagene locus. Curr. Top. Microbiol. Immunol. 1997, 224, 67–72. [Google Scholar]
- Nagoshi, H.; Taki, T.; Hanamura, I.; Nitta, M.; Otsuki, T.; Nishida, K.; Okuda, K.; Sakamoto, N.; Kobayashi, S.; Yamamoto-Sugitani, M.; et al. Frequent PVT1 rearrangement and novel chimeric genes PVT1–NBEA and PVT1–WWOX occur in multiple myeloma with 8q24 abnormality. Cancer Res. 2012, 72, 4954–4962. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Tseng, Y.-Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 1–19. [Google Scholar] [CrossRef]
- Cui, M.; You, L.; Ren, X.; Zhao, W.; Liao, Q.; Zhao, Y. Biochemical and Biophysical Research Communications. Biochem. Biophys. Res. Commun. 2016, 471, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Kuo, W.-L.; Stilwell, J.L.; Takano, H.; Lapuk, A.V.; Fridlyand, J.; Mao, J.-H.; Yu, M.; Miller, M.A.; Santos, J.L.; et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 2007, 13, 5745–5755. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, E.; Suraokar, M.B.; Rodriguez, J.; Mino, B.; Lin, H.Y.; Rice, D.C.; Tsao, A.; Wistuba, I.I. Frequent coamplification and cooperation between C-MYC and PVT1 oncogenes promote malignant pleural mesothelioma. J. Thorac. Oncol. 2014, 9, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, D.; Gong, M.; Wang, J.; Huang, X.; Wu, T.; Wang, C. Expression and clinical significance of the long non-coding RNA PVT1 in human gastric cancer. Onco Targets 2014, 7, 1625–1630. [Google Scholar] [CrossRef]
- Yang, Y.-R.; Zang, S.-Z.; Zhong, C.-L.; Li, Y.-X.; Zhao, S.-S.; Feng, X.-J. Increased expression of the lncRNA PVT1 promotes tumorigenesis in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 6929–6935. [Google Scholar]
- Huang, C.; Yu, W.; Wang, Q.; Cui, H.; Wang, Y.; Zhang, L.; Han, F.; Huang, T. Increased expression of the lncRNA PVT1 is associated with poor prognosis in pancreatic cancer patients. Minerva Med. 2015, 106, 143–149. [Google Scholar]
- Zeng, C.; Yu, X.; Lai, J.; Yang, L.; Chen, S.; Li, Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 2015, 8, 126. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chen, W.; Iyer, M.K.; Cao, Q.; Ma, T.; Han, S.; Sahu, A.; Malik, R.; Wilder-Romans, K.; Navone, N.; et al. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014, 74, 1651–1660. [Google Scholar] [CrossRef]
- Xiang, J.-F.; Yin, Q.-F.; Chen, T.; Zhang, Y.; Zhang, X.-O.; Wu, Z.; Zhang, S.; Wang, H.-B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Nat. Publ. Group 2014, 24, 513–531. [Google Scholar]
- Chen, L.; Wang, W.; Cao, L.; Li, Z.; Wang, X. Long Non-Coding RNA CCAT1 Acts as a Competing Endogenous RNA to Regulate Cell Growth and Differentiation in Acute Myeloid Leukemia. Mol. Cells 2016, 39, 330–336. [Google Scholar]
- Chen, S.; Gu, T.; Lu, Z.; Qiu, L.; Xiao, G.; Zhu, X.; Li, F.; Yu, H.; Li, G.; Liu, H. Roles of MYC-targeting long non-coding RNA MINCR in cell cycle regulation and apoptosis in non-small cell lung Cancer. Respir. Res. 2019, 20, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, G.; Hu, X.; Wu, L.; Feng, Y.; He, S.; Zhang, Y.; Hu, Z.; Yang, L.; Tian, T.; et al. Oncogenic RAS regulates long non-coding RNA Orilnc1 in human cancer. Cancer Res. 2017, 77, 3745–3757. [Google Scholar] [CrossRef] [PubMed]
- Esposito, R.; Esposito, D.; Pallante, P.; Fusco, A.; Ciccodicola, A.; Costa, V. Oncogenic Properties of the Antisense lncRNA COMETin BRAF- and RET-Driven Papillary Thyroid Carcinomas. Cancer Res. 2019, 79, 2124–2135. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Vucic, E.A.; Enfield, K.S.S.; Stewart, G.L.; Lonergan, K.M.; Kennett, J.Y.; Becker-Santos, D.D.; MacAulay, C.E.; Lam, S.; Brown, C.J.; et al. Human cancer long non-coding RNA transcriptomes. PLoS ONE 2011, 6, e25915. [Google Scholar] [CrossRef]
- Hudson, W.H.; Pickard, M.R.; de Vera, I.M.S.; Kuiper, E.G.; Mourtada-Maarabouni, M.; Conn, G.L.; Kojetin, D.J.; Williams, G.T.; Ortlund, E.A. Conserved sequence-specific lincRNA-steroid receptor interactions drive transcriptional repression and direct cell fate. Nat. Commun. 2014, 5, 5395. [Google Scholar] [CrossRef]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef]
- Schneider, C.; King, R.M.; Philipson, L. Genes specifically expressed at growth arrest of mammalian cells. Cell 1988, 54, 787–793. [Google Scholar] [CrossRef]
- Mourtada-Maarabouni, M.; Pickard, M.R.; Hedge, V.L.; Farzaneh, F.; Williams, G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Nat. Publ. Group 2009, 28, 195–208. [Google Scholar] [CrossRef]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef]
- Pickard, M.R.; Williams, G.T. Molecular and Cellular Mechanisms of Action of Tumour Suppressor GAS5 LncRNA. Genes 2015, 6, 484–499. [Google Scholar] [CrossRef]
- Goustin, A.S.; Thepsuwan, P.; Kosir, M.A.; Lipovich, L. The Growth-Arrest-Specific (GAS)-5 Long Non-Coding RNA: A Fascinating lncRNA Widely Expressed in Cancers. Noncoding RNA 2019, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Schuster-Gossler, K.; Bilinski, P.; Sado, T.; Ferguson-Smith, A.; Gossler, A. The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an RNA. Dev. Dyn 1998, 212, 214–228. [Google Scholar] [CrossRef]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000, 5, 211–220. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted Monocytic miR-150 Enhances Targeted Endothelial Cell Migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef]
- Braconi, C.; Kogure, T.; Valeri, N.; Huang, N.; Nuovo, G.; Costinean, S.; Negrini, M.; Miotto, E.; Croce, C.M.; Patel, T. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Nat. Publ. Group 2011, 30, 4750–4756. [Google Scholar] [CrossRef]
- Wang, P.; Ren, Z.; Sun, P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J. Cell. Biochem. 2012, 113, 1868–1874. [Google Scholar] [CrossRef]
- Lyu, Y.; Lou, J.; Yang, Y.; Feng, J.; Hao, Y.; Huang, S.; Yin, L.; Xu, J.; Huang, D.; Ma, B.; et al. Dysfunction of the WT1-MEG3 signaling promotes AML leukemogenesis via p53-dependent and -independent pathways. Leukemia 2017, 31, 2543–2551. [Google Scholar] [CrossRef]
- Li, J.; Zi, Y.; Wang, W.; Li, Y. Long Noncoding RNA MEG3 Inhibits Cell Proliferation and Metastasis in Chronic Myeloid Leukemia via Targeting miR-184. Oncol Res. 2018, 26, 297–305. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Wagner, K.; Gentle, D.; Cooper, W.N.; Catchpoole, D.; Grundy, R.; Ferguson-Smith, A.C.; Maher, E.R. Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: Analysis of neuroblastoma, phaeochromocytoma and Wilms’ tumour. Br. J. Cancer 2005, 92, 1574–1580. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Klibanski, A. MEG3 noncoding RNA: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, R45–R53. [Google Scholar] [CrossRef] [PubMed]
- Gou, Q.; Gao, L.; Nie, X.; Pu, W.; Zhu, J.; Wang, Y.; Liu, X.; Tan, S.; Zhou, J.-K.; Gong, Y.; et al. Long Noncoding RNA AB074169 Inhibits Cell Proliferation via Modulation of KHSRP-Mediated CDKN1a Expression in Papillary Thyroid Carcinoma. Cancer Res. 2018, 78, 4163–4174. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kopp, F.; Chang, T.-C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Tichon, A.; Gil, N.; Lubelsky, Y.; Havkin Solomon, T.; Lemze, D.; Itzkovitz, S.; Stern-Ginossar, N.; Ulitsky, I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 2016, 7, 12209–12210. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Zamudio, J.R.; Jong, R.M.; Soukup, D.; Resnick, R.; Sarma, K.; Ward, A.J.; Raj, A.; Lee, J.T.; Sharp, P.A.; et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol. Cell 2014, 54, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Fesler, A.; Schee, K.; Fodstad, O.; Flatmark, K.; Ju, J. Clinical significance of long intergenic noncoding RNA-p21 in colorectal cancer. Clin. Colorectal. Cancer 2013, 12, 261–266. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, H.; Mei, Y.; Wu, M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Mol. Cell 2014, 53, 88–100. [Google Scholar] [CrossRef]
- Doucrasy, S.; Coll, J.; Barrois, M.; Joubel, A.; Prost, S.; Dozier, C.; Stehelin, D.; Riou, G. Expression of the human fetal bac h19 gene in invasive cancers. Int. J. Oncol. 1993, 2, 753–758. [Google Scholar] [CrossRef]
- Berteaux, N.; Lottin, S.; Monté, D.; Pinte, S.; Quatannens, B.; Coll, J.; Hondermarck, H.; Curgy, J.-J.; Dugimont, T.; Adriaenssens, E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem. 2005, 280, 29625–29636. [Google Scholar] [CrossRef]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Dugimont, T.; Montpellier, C.; Adriaenssens, E.; Lottin, S.; Dumont, L.; Iotsova, V.; Lagrou, C.; Stehelin, D.; Coll, J.; Curgy, J.J. The H19 TATA-less promoter is efficiently repressed by wild-type tumor suppressor gene product p53. Oncogene 1998, 16, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Barsyte-Lovejoy, D.; Lau, S.K.; Boutros, P.C.; Khosravi, F.; Jurisica, I.; Andrulis, I.L.; Tsao, M.S.; Penn, L.Z. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006, 66, 5330–5337. [Google Scholar] [CrossRef]
- Matouk, I.J.; Mezan, S.; Mizrahi, A.; Ohana, P.; Abu-Lail, R.; Fellig, Y.; Degroot, N.; Galun, E.; Hochberg, A. The oncofetal H19 RNA connection: Hypoxia, p53 and cancer. Biochim. Biophys. Acta 2010, 1803, 443–451. [Google Scholar] [CrossRef]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rüger, R. Long Non-coding RNAs and their Role in Metastasis. Cancer Genom. Proteom. 2017, 14, 143–160. [Google Scholar] [CrossRef]
- Hao, Y.; Crenshaw, T.; Moulton, T.; Newcomb, E.; Tycko, B. Tumour-suppressor activity of H19 RNA. Nature 1993, 365, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Yoshimizu, T.; Miroglio, A.; Ripoche, M.-A.; Gabory, A.; Vernucci, M.; Riccio, A.; Colnot, S.; Godard, C.; Terris, B.; Jammes, H.; et al. The H19 locus acts in vivo as a tumor suppressor. Proc. Natl. Acad. Sci. USA 2008, 105, 12417–12422. [Google Scholar] [CrossRef]
- Gabory, A.; Ripoche, M.-A.; Le Digarcher, A.; Watrin, F.; Ziyyat, A.; Forné, T.; Jammes, H.; Ainscough, J.F.X.; Surani, M.A.; Journot, L.; et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 2009, 136, 3413–3421. [Google Scholar] [CrossRef]
- Kallen, A.N.; Zhou, X.-B.; Xu, J.; Qiao, C.; Ma, J.; Yan, L.; Lu, L.; Liu, C.; Yi, J.-S.; Zhang, H.; et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell 2013, 52, 101–112. [Google Scholar] [CrossRef]
- Tsang, W.P.; Ng, E.K.O.; Ng, S.S.M.; Jin, H.; Yu, J.; Sung, J.J.Y.; Kwok, T.T. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010, 31, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef]
- Rheinbay, E.; Parasuraman, P.; Grimsby, J.; Tiao, G.; Engreitz, J.M.; Kim, J.; Lawrence, M.S.; Taylor-Weiner, A.; Rodriguez-Cuevas, S.; Rosenberg, M.; et al. Recurrent and functional regulatory mutations in breast cancer. Nature 2017, 547, 55–60. [Google Scholar] [PubMed]
- Knutsen, E.; Lellahi, S.M.; Aure, M.R.; Nord, S.; Fismen, S.; Larsen, K.B.; Gabriel, M.T.; Hedberg, A.; Bjørklund, S.S.; Oslo Breast Cancer Research Consortium (OSBREAC); et al. The expression of the long NEAT1_2 isoform is associated with human epidermal growth factor receptor 2-positive breast cancers. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Sinow, C.; Raj, N.; Mazur, P.K.; Bieging-Rolett, K.; Broz, D.K.; Imam, J.F.C.; Vogel, H.; Wood, L.D.; Sage, J.; et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Xu, Y.; Xu, L.; Yu, X.; Cheng, J.; Yang, L.; Chen, S.; Li, Y. Inhibition of long non-coding RNA NEAT1 impairs myeloid differentiation in acute promyelocytic leukemia cells. BMC Cancer 2014, 14, 693. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, S.; Zhao, Y.; Du, F.; Wang, W.; Lv, P.; Qi, L. Long noncoding RNA NEAT1 modulates cell proliferation and apoptosis by regulating miR-23a-3p/SMC1A in acute myeloid leukemia. J. Cell Physiol. 2019, 234, 6161–6172. [Google Scholar] [CrossRef]
- Flockhart, R.J.; Webster, D.E.; Qu, K.; Mascarenhas, N.; Kovalski, J.; Kretz, M.; Khavari, P.A. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res. 2012, 22, 1006–1014. [Google Scholar] [CrossRef]
- Li, R.; Zhang, L.; Jia, L.; Duan, Y.; Li, Y.; Bao, L.; Sha, N. Long non-coding RNA BANCR promotes proliferation in malignant melanoma by regulating MAPK pathway activation. PLoS ONE 2014, 9, e100893. [Google Scholar] [CrossRef]
- Sun, M.; Liu, X.-H.; Wang, K.-M.; Nie, F.-Q.; Kong, R.; Yang, J.-S.; Xia, R.; Xu, T.-P.; Jin, F.-Y.; Liu, Z.-J.; et al. Downregulation of BRAF activated non-coding RNA is associated with poor prognosis for non-small cell lung cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Mol. Cancer 2014, 13, 68. [Google Scholar] [CrossRef]
- Xue, S.; Jiang, S.-Q.; Li, Q.-W.; Wang, S.; Li, J.; Yang, S.; Zhang, H.-M.; Xu, Y.-F.; Wang, L.-S.; Zheng, J.-H. Decreased expression of BRAF-activated long non-coding RNA is associated with the proliferation of clear cell renal cell carcinoma. BMC Urol. 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Goyal, A.; Myacheva, K.; Gross, M.; Klingenberg, M.; Duran Arqué, B.; Diederichs, S. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017, 45, e12. [Google Scholar] [CrossRef]
- Awwad, D.A. Beyond classic editing: Innovative CRISPR approaches for functional studies of long non-coding RNA. Biol. Methods Protoc. 2019, 4, bpz017. [Google Scholar] [PubMed]
- Han, J.; Zhang, J.; Chen, L.; Shen, B.; Zhou, J.; Hu, B.; Du, Y.; Tate, P.H.; Huang, X.; Zhang, W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014, 11, 829–835. [Google Scholar] [CrossRef]
- Yin, Y.; Yan, P.; Lu, J.; Song, G.; Zhu, Y.; Li, Z.; Zhao, Y.; Shen, B.; Huang, X.; Zhu, H.; et al. Opposing Roles for the lncRNA Haunt and Its Genomic Locus in Regulating HOXA Gene Activation during Embryonic Stem Cell Differentiation. Cell Stem Cell 2015, 16, 504–516. [Google Scholar] [CrossRef]
- Aparicio-Prat, E.; Arnan, C.; Sala, I.; Bosch, N.; Guigó, R.; Johnson, R. DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genom. 2016, 16, 1–15. [Google Scholar]
- Esposito, R.; Bosch, N.; Lanzós, A.; Polidori, T.; Pulido-Quetglas, C.; Johnson, R. Hacking the Cancer Genome: Profiling Therapeutically Actionable Long Non-coding RNAs Using CRISPR-Cas9 Screening. Cancer Cell 2019, 35, 545–557. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Koch, L. Functional genomics: Screening for lncRNA function. Nat. Rev. Genet. 2017, 18, 70. [Google Scholar]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.-H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, X.; Yuan, J.; Geng, T.; Chen, S.; Hu, X.; Cui, I.H.; Cui, H. Biallelic insertion of a transcriptional terminator via the CRISPR/Cas9 system efficiently silences expression of protein-coding and non-coding RNA genes. J. Biol. Chem. 2017, 292, 5624–5633. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, G.; Li, J.; Zhang, X.; Huang, S.; Xiang, S.; Hu, X.; Liu, C. CRISPRlnc: A manually curated database of validated sgRNAs for lncRNAs. Nucleic Acids Res. 2019, 47, D63–D68. [Google Scholar] [CrossRef]
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef]
- Janga, H.; Aznaourova, M.; Boldt, F.; Damm, K.; Grünweller, A.; Schulte, L.N. Cas9-mediated excision of proximal DNaseI/H3K4me3 signatures confers robust silencing of microRNA and long non-coding RNA genes. PLoS ONE 2018, 13, e0193066. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Höning, K.; Ebert, T.S.; Hornung, V. CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Cheng, T.-L.; Qiu, Z. Long non-coding RNA tagging and expression manipulation via CRISPR/Cas9-mediated targeted insertion. Protein Cell 2018, 9, 820–825. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-Z.; Wang, Y.; Li, S.-Q.; Yao, R.-W.; Luan, P.-F.; Wu, H.; Carmichael, G.G.; Chen, L.-L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997.e7. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Cai, Y.; Tang, L.; Han, X.; Gao, F.; Cao, H.; Qi, F.; Kapranov, P. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 2020, 10, 1794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; You, Y. CRISPR-Cas13a system: A novel approach to precision oncology. Cancer Biol. Med. 2020, 17, 6–8. [Google Scholar]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef]
- Hannon, G.J.; Rossi, J.J. Unlocking the potential of the human genome with RNA interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, N.; Gyntelberg, F.; Faber, J. The appraisal of chronic stress and the development of the metabolic syndrome: A systematic review of prospective cohort studies. Endocr. Connect. 2014, 3, R55–R80. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221. [Google Scholar] [CrossRef]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2’-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef]
- Yoo, B.H.; Bochkareva, E.; Bochkarev, A.; Mou, T.-C.; Gray, D.M. 2’-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004, 32, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Lagendijk, A.K.; Ketting, R.F.; Moulton, J.D.; Plasterk, R.H.A. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. PLoS Biol. 2007, 5, e203. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Geary, R.S.; Baker, B.F.; Crooke, S.T. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (kynamro(®)): A second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin. Pharm. 2015, 54, 133–146. [Google Scholar] [CrossRef]
- Anthony, K.; Feng, L.; Arechavala-Gomeza, V.; Guglieri, M.; Straub, V.; Bushby, K.; Cirak, S.; Morgan, J.; Muntoni, F. Exon skipping quantification by quantitative reverse-transcription polymerase chain reaction in Duchenne muscular dystrophy patients treated with the antisense oligomer eteplirsen. Hum. Gene Methods 2012, 23, 336–345. [Google Scholar] [CrossRef]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Moulton, J.D. Using Morpholinos to Control Gene Expression. Curr. Protoc. Nucleic Acid Chem. 2017, 68, 4.30.1–4.30.29. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Janson, A.A.M.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.-J.B.; van Deutekom, J.C.T. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Staton, A.A.; Giraldez, A.J. Use of target protector morpholinos to analyze the physiological roles of specific miRNA-mRNA pairs in vivo. Nat. Protoc. 2011, 6, 2035–2049. [Google Scholar] [CrossRef]
- Blum, M.; De Robertis, E.M.; Wallingford, J.B.; Niehrs, C. Morpholinos: Antisense and Sensibility. Dev. Cell 2015, 35, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Pegueroles, C.; Gabaldón, T. Secondary structure impacts patterns of selection in human lncRNAs. BMC Biol. 2016, 14, 1–13. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef]
- Howe, J.A.; Wang, H.; Fischmann, T.O.; Balibar, C.J.; Xiao, L.; Galgoci, A.M.; Malinverni, J.C.; Mayhood, T.; Villafania, A.; Nahvi, A.; et al. Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526, 672–677. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Senn, J.J.; Burel, S.; Henry, S.P. Non-CpG-containing antisense 2’-methoxyethyl oligonucleotides activate a proinflammatory response independent of Toll-like receptor 9 or myeloid differentiation factor 88. J. Pharm. Exp. 2005, 314, 972–979. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Vollmer, J.; Weeratna, R.D.; Jurk, M.; Samulowitz, U.; McCluskie, M.J.; Payette, P.; Davis, H.L.; Schetter, C.; Krieg, A.M. Oligodeoxynucleotides lacking CpG dinucleotides mediate Toll-like receptor 9 dependent T helper type 2 biased immune stimulation. Immunology 2004, 113, 212–223. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Bhagat, L.; Wang, D.; Yu, D.; Sullivan, T.; La Monica, N.; Agrawal, S. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 2013, 41, 3947–3961. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aprile, M.; Katopodi, V.; Leucci, E.; Costa, V. LncRNAs in Cancer: From garbage to Junk. Cancers 2020, 12, 3220. https://doi.org/10.3390/cancers12113220
Aprile M, Katopodi V, Leucci E, Costa V. LncRNAs in Cancer: From garbage to Junk. Cancers. 2020; 12(11):3220. https://doi.org/10.3390/cancers12113220
Chicago/Turabian StyleAprile, Marianna, Vicky Katopodi, Eleonora Leucci, and Valerio Costa. 2020. "LncRNAs in Cancer: From garbage to Junk" Cancers 12, no. 11: 3220. https://doi.org/10.3390/cancers12113220
APA StyleAprile, M., Katopodi, V., Leucci, E., & Costa, V. (2020). LncRNAs in Cancer: From garbage to Junk. Cancers, 12(11), 3220. https://doi.org/10.3390/cancers12113220