Thyroid Cancer and Circadian Clock Disruption

 ,
,  ,
,  ,
,  ,
,  and
and 
Simple Summary
Abstract
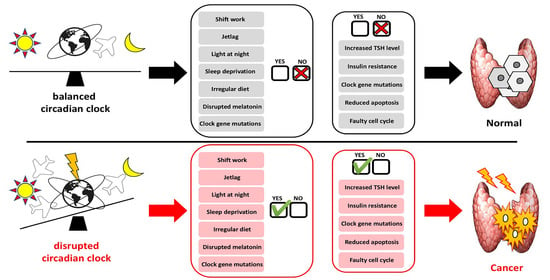
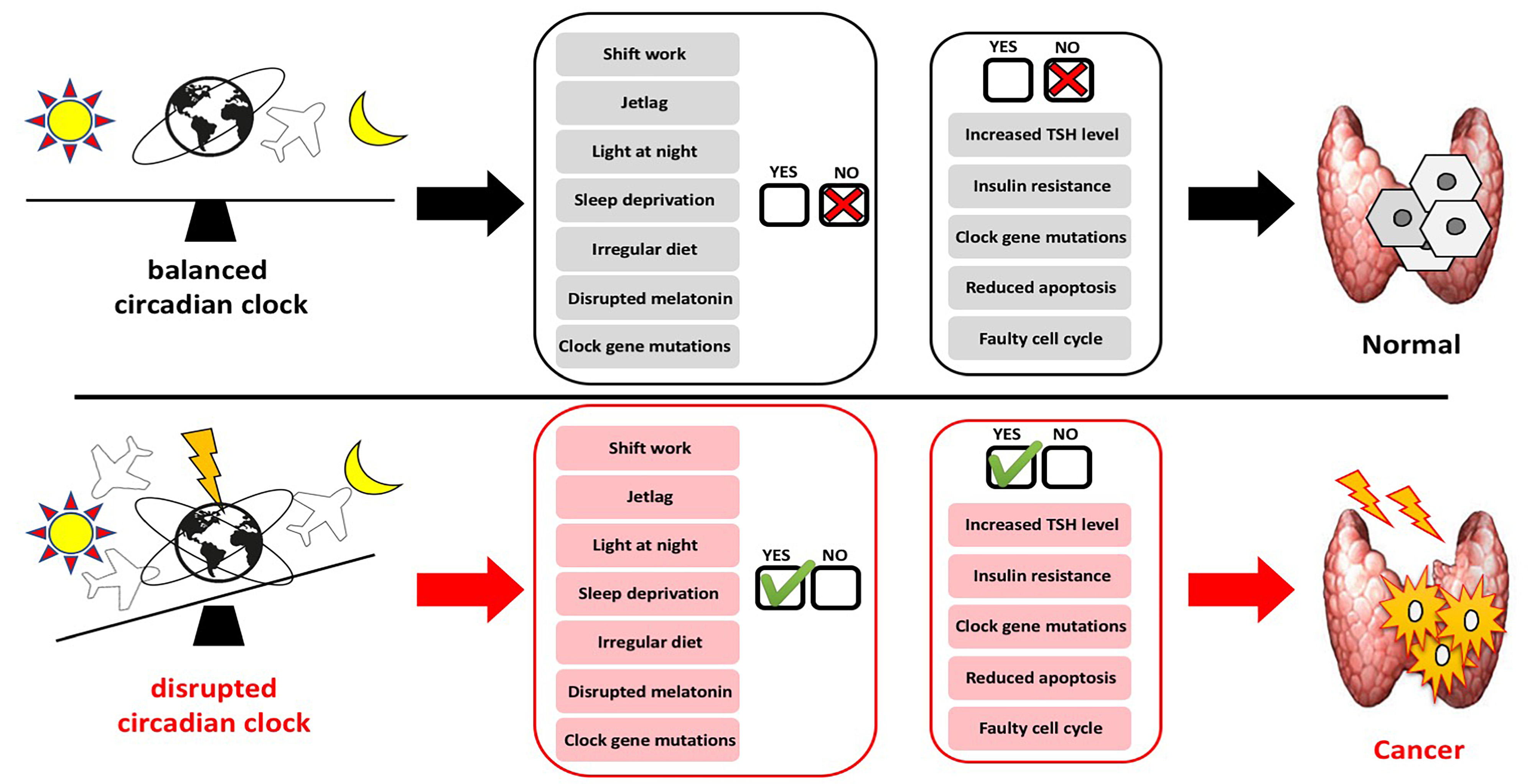
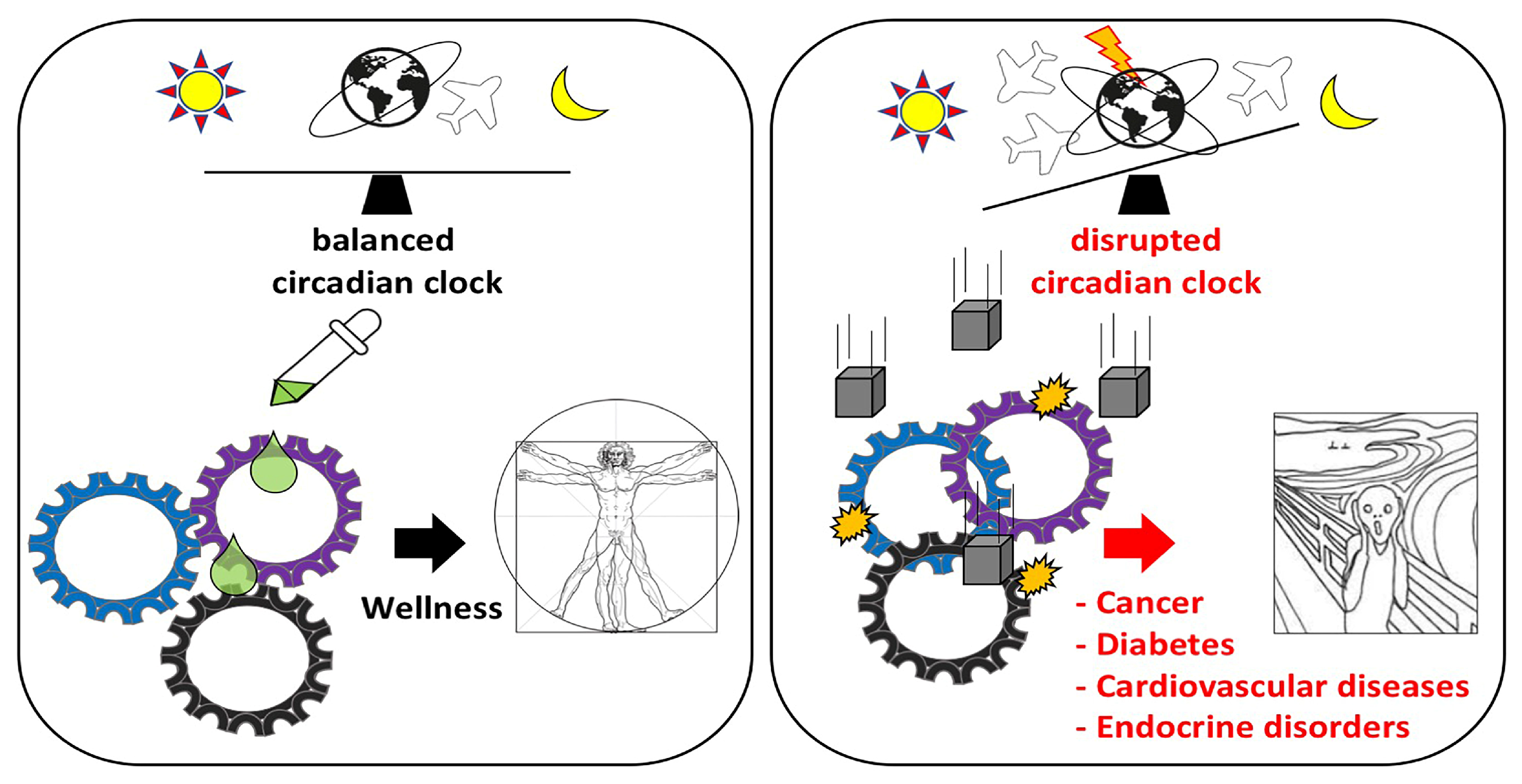
1. Introduction
2. Circadian Clock
2.1. Regulation
2.2. Circadian Clock and Cell Cycle
2.3. Tumor Suppressor or Oncogene: The Janus Face of the Circadian Clock Machinery
2.4. Circadian Clock and Stemness
3. Thyroid Tumorigenesis
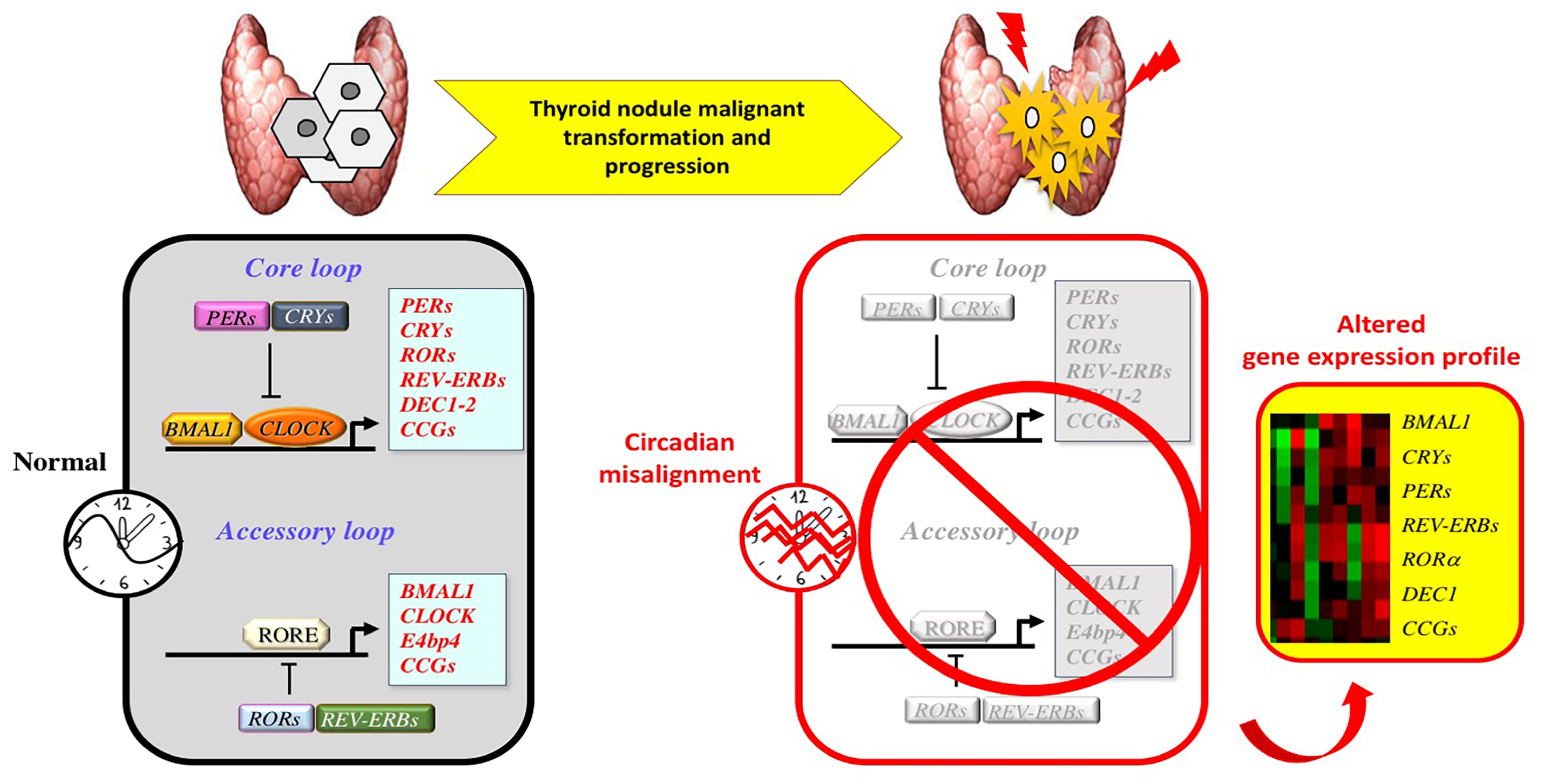
4. Circadian Clock and Thyroid Tumorigenesis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATC | Anaplastic Thyroid Cancer |
| ATM | Ataxia Telangiectasia Mutated |
| ATR | Ataxia Telangiectasia and Rad3-related protein |
| CCGs | Clock-Controlled Genes |
| CCSC | Colon Cancer Stem-like Cells |
| DBP | D-box-binding protein |
| DDR1 | Discoidin Domain Receptor 1 |
| DEC1 | Differentially Expressed in Chondrocyte 1 |
| DKO | Double knockout |
| ES | Embryonic stem cells |
| FNA | Fine-Needle Aspiration |
| FTC | Follicular Thyroid Cancer |
| GSC | Glioma Stem Cells |
| HAT | Histone acetyltransferase |
| HCC | Hurthle Cell Carcinoma |
| HGF | Hepatocyte Growth Factor |
| HPT | Hypothalamic-Pituitary-Thyroid |
| IGF-1R | Insulin-like growth factor-1 receptor |
| IGF-2 | Insulin-like growth factor-2 |
| IGF | Insulin-like growth factor |
| KO | Knockout |
| lncRNAs | Long-non coding RNAs |
| miRNAs | MicroRNAs |
| PAX8/PPAR | Paired-box gene 8/Peroxisome Proliferator-Activated Receptor gamma |
| PDTC | Poorly Differentiated Thyroid Cancer |
| PKB | Protein Kinase B |
| PTC | Papillary Thyroid Cancer |
| SCN | Suprachiasmatic Nucleus |
| TC | Thyroid Cancer |
| TCSC | Thyroid Cancer Stem Cell |
| TSH | Thyroid-Stimulating Hormone |
References
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Vella, V.; Nicolosi, M.L.; Belfiore, A. Insulin Resistance: Any Role in the Changing Epidemiology of Thyroid Cancer? Front. Endocrinol. 2017, 8, 314. [Google Scholar] [CrossRef]
- Chitikova, Z.; Pusztaszeri, M.; Makhlouf, A.M.; Berczy, M.; Delucinge-Vivier, C.; Triponez, F.; Meyer, P.; Philippe, J.; Dibner, C. Identification of new biomarkers for human papillary thyroid carcinoma employing NanoString analysis. Oncotarget 2015, 6, 10978–10993. [Google Scholar] [CrossRef]
- Ikegami, K.; Refetoff, S.; Van Cauter, E.; Yoshimura, T. Interconnection between circadian clocks and thyroid function. Nat. Rev. Endocrinol. 2019, 15, 590–600. [Google Scholar] [CrossRef]
- Makhlouf, A.M.; Chitikova, Z.; Pusztaszeri, M.; Berczy, M.; Delucinge-Vivier, C.; Triponez, F.; Meyer, P.; Philippe, J.; Dibner, C. Identification of CHEK1, SLC26A4, c-KIT, TPO and TG as new biomarkers for human follicular thyroid carcinoma. Oncotarget 2016, 7, 45776–45788. [Google Scholar] [CrossRef]
- Mannic, T.; Meyer, P.; Triponez, F.; Pusztaszeri, M.; Le Martelot, G.; Mariani, O.; Schmitter, D.; Sage, D.; Philippe, J.; Dibner, C. Circadian clock characteristics are altered in human thyroid malignant nodules. J. Clin. Endocrinol. Metab. 2013, 98, 4446–4456. [Google Scholar] [CrossRef]
- Philippe, J.; Dibner, C. Thyroid circadian timing: Roles in physiology and thyroid malignancies. J. Biol. Rhythms 2015, 30, 76–83. [Google Scholar] [CrossRef]
- Angelousi, A.; Kassi, E.; Ansari-Nasiri, N.; Randeva, H.; Kaltsas, G.; Chrousos, G. Clock genes and cancer development in particular in endocrine tissues. Endocr. Relat. Cancer 2019, 26, R305–R317. [Google Scholar] [CrossRef]
- Turek, F.W. Circadian clocks: Not your grandfather’s clock. Science 2016, 354, 992–993. [Google Scholar] [CrossRef] [PubMed]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Buxton, O.M.; Cain, S.W.; O’Connor, S.P.; Porter, J.H.; Duffy, J.F.; Wang, W.; Czeisler, C.A.; Shea, S.A. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci. Transl. Med. 2012, 4, 129ra143. [Google Scholar] [CrossRef] [PubMed]
- Kettner, N.M.; Katchy, C.A.; Fu, L. Circadian gene variants in cancer. Ann. Med. 2014, 46, 208–220. [Google Scholar] [CrossRef]
- Scheer, F.A.; Hilton, M.F.; Mantzoros, C.S.; Shea, S.A. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl. Acad. Sci. USA 2009, 106, 4453–4458. [Google Scholar] [CrossRef]
- He, B.; Nohara, K.; Park, N.; Park, Y.S.; Guillory, B.; Zhao, Z.; Garcia, J.M.; Koike, N.; Lee, C.C.; Takahashi, J.S.; et al. The Small Molecule Nobiletin Targets the Molecular Oscillator to Enhance Circadian Rhythms and Protect against Metabolic Syndrome. Cell Metab. 2016, 23, 610–621. [Google Scholar] [CrossRef]
- Winter, C.; Silvestre-Roig, C.; Ortega-Gomez, A.; Lemnitzer, P.; Poelman, H.; Schumski, A.; Winter, J.; Drechsler, M.; de Jong, R.; Immler, R.; et al. Chrono-pharmacological Targeting of the CCL2-CCR2 Axis Ameliorates Atherosclerosis. Cell Metab. 2018, 28, 175–182 e175. [Google Scholar] [CrossRef]
- Boelaert, K.; Horacek, J.; Holder, R.L.; Watkinson, J.C.; Sheppard, M.C.; Franklyn, J.A. Serum thyrotropin concentration as a novel predictor of malignancy in thyroid nodules investigated by fine-needle aspiration. J. Clin. Endocrinol. Metab. 2006, 91, 4295–4301. [Google Scholar] [CrossRef]
- Haymart, M.R.; Repplinger, D.J.; Leverson, G.E.; Elson, D.F.; Sippel, R.S.; Jaume, J.C.; Chen, H. Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage. J. Clin. Endocrinol. Metab. 2008, 93, 809–814. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kita, M.; Efstathiadou, Z.; Poulakos, P.; Slavakis, A.; Sofianou, D.; Flaris, N.; Leontsini, M.; Kourtis, A.; Avramidis, A. Serum thyrotropin concentration as a biochemical predictor of thyroid malignancy in patients presenting with thyroid nodules. J. Cancer Res. Clin. Oncol. 2008, 134, 953–960. [Google Scholar] [CrossRef]
- Moon, S.H.; Lee, B.J.; Kim, S.J.; Kim, H.C. Relationship between thyroid stimulating hormone and night shift work. Ann. Occup. Environ. Med. 2016, 28, 53. [Google Scholar] [CrossRef] [PubMed]
- Bellastella, A.; Pisano, G.; Iorio, S.; Pasquali, D.; Orio, F.; Venditto, T.; Sinisi, A.A. Endocrine secretions under abnormal light-dark cycles and in the blind. Horm. Res. 1998, 49, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, K.; Leproult, R.; Van Cauter, E. Impact of sleep debt on metabolic and endocrine function. Lancet 1999, 354, 1435–1439. [Google Scholar] [CrossRef]
- Cibas, E.S.; Ali, S.Z. The Bethesda System for Reporting Thyroid Cytopathology. Thyroid 2009, 19, 1159–1165. [Google Scholar] [CrossRef]
- Baloch, Z.W.; Fleisher, S.; LiVolsi, V.A.; Gupta, P.K. Diagnosis of “follicular neoplasm”: A gray zone in thyroid fine-needle aspiration cytology. Diagn. Cytopathol. 2002, 26, 41–44. [Google Scholar] [CrossRef]
- Walsh, P.S.; Wilde, J.I.; Tom, E.Y.; Reynolds, J.D.; Chen, D.C.; Chudova, D.I.; Pagan, M.; Pankratz, D.G.; Wong, M.; Veitch, J.; et al. Analytical performance verification of a molecular diagnostic for cytology-indeterminate thyroid nodules. J. Clin. Endocrinol. Metab. 2012, 97, E2297–E2306. [Google Scholar] [CrossRef]
- Yang, G.C.; Liebeskind, D.; Messina, A.V. Should cytopathologists stop reporting follicular neoplasms on fine-needle aspiration of the thyroid? Cancer 2003, 99, 69–74. [Google Scholar] [CrossRef]
- Yang, J.; Schnadig, V.; Logrono, R.; Wasserman, P.G. Fine-needle aspiration of thyroid nodules: A study of 4703 patients with histologic and clinical correlations. Cancer 2007, 111, 306–315. [Google Scholar] [CrossRef]
- Yassa, L.; Cibas, E.S.; Benson, C.B.; Frates, M.C.; Doubilet, P.M.; Gawande, A.A.; Moore, F.D., Jr.; Kim, B.W.; Nose, V.; Marqusee, E.; et al. Long-term assessment of a multidisciplinary approach to thyroid nodule diagnostic evaluation. Cancer 2007, 111, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Eszlinger, M.; Lau, L.; Ghaznavi, S.; Symonds, C.; Chandarana, S.P.; Khalil, M.; Paschke, R. Molecular profiling of thyroid nodule fine-needle aspiration cytology. Nat. Rev. Endocrinol. 2017, 13, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Park, Y.J. Genomic Characterization of Differentiated Thyroid Carcinoma. Endocrinol. Metab. 2019, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Liu, R.; Liu, X.; Murugan, A.K.; Zhu, G.; Zeiger, M.A.; Pai, S.; Bishop, J. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J. Clin. Oncol. 2014, 32, 2718–2726. [Google Scholar] [CrossRef] [PubMed]
- Grani, G.; Sponziello, M.; Pecce, V.; Ramundo, V.; Durante, C. Contemporary Thyroid Nodule Evaluation and Management. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Mercurio, S.; Wald, A.I.; Barbi de Moura, M.; Callenberg, K.; Santana-Santos, L.; Gooding, W.E.; Yip, L.; Ferris, R.L.; Nikiforov, Y.E. Analytical performance of the ThyroSeq v3 genomic classifier for cancer diagnosis in thyroid nodules. Cancer 2018, 124, 1682–1690. [Google Scholar] [CrossRef]
- Simon, R.; Radmacher, M.D.; Dobbin, K.; McShane, L.M. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J. Natl. Cancer Inst. 2003, 95, 14–18. [Google Scholar] [CrossRef]
- Moore, R.Y.; Eichler, V.B. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972, 42, 201–206. [Google Scholar] [CrossRef]
- Ralph, M.R.; Foster, R.G.; Davis, F.C.; Menaker, M. Transplanted suprachiasmatic nucleus determines circadian period. Science 1990, 247, 975–978. [Google Scholar] [CrossRef]
- Herzog, E.D.; Takahashi, J.S.; Block, G.D. Clock controls circadian period in isolated suprachiasmatic nucleus neurons. Nat. Neurosci. 1998, 1, 708–713. [Google Scholar] [CrossRef]
- Shearman, L.P.; Sriram, S.; Weaver, D.R.; Maywood, E.S.; Chaves, I.; Zheng, B.; Kume, K.; Lee, C.C.; van der Horst, G.T.; Hastings, M.H.; et al. Interacting molecular loops in the mammalian circadian clock. Science 2000, 288, 1013–1019. [Google Scholar] [CrossRef]
- Harmer, S.L.; Panda, S.; Kay, S.A. Molecular bases of circadian rhythms. Annu. Rev. Cell Dev. Biol. 2001, 17, 215–253. [Google Scholar] [CrossRef] [PubMed]
- Darlington, T.K.; Wager-Smith, K.; Ceriani, M.F.; Staknis, D.; Gekakis, N.; Steeves, T.D.; Weitz, C.J.; Takahashi, J.S.; Kay, S.A. Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science 1998, 280, 1599–1603. [Google Scholar] [CrossRef]
- Wickwire, E.M.; Geiger-Brown, J.; Scharf, S.M.; Drake, C.L. Shift Work and Shift Work Sleep Disorder: Clinical and Organizational Perspectives. Chest 2017, 151, 1156–1172. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.H. Circadian clockwork: Two loops are better than one. Nat. Rev. Neurosci. 2000, 1, 143–146. [Google Scholar] [CrossRef] [PubMed]
- King, D.P.; Takahashi, J.S. Molecular genetics of circadian rhythms in mammals. Annu. Rev. Neurosci. 2000, 23, 713–742. [Google Scholar] [CrossRef] [PubMed]
- Lowrey, P.L.; Takahashi, J.S. Genetics of the mammalian circadian system: Photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Annu. Rev. Genet. 2000, 34, 533–562. [Google Scholar] [CrossRef]
- Reppert, S.M.; Weaver, D.R. Molecular analysis of mammalian circadian rhythms. Annu. Rev. Physiol. 2001, 63, 647–676. [Google Scholar] [CrossRef]
- Sato, F.; Kawamoto, T.; Fujimoto, K.; Noshiro, M.; Honda, K.K.; Honma, S.; Honma, K.; Kato, Y. Functional analysis of the basic helix-loop-helix transcription factor DEC1 in circadian regulation. Interaction with BMAL1. Eur. J. Biochem. 2004, 271, 4409–4419. [Google Scholar] [CrossRef]
- Eide, E.J.; Kang, H.; Crapo, S.; Gallego, M.; Virshup, D.M. Casein kinase I in the mammalian circadian clock. Methods Enzymol. 2005, 393, 408–418. [Google Scholar] [CrossRef]
- Lee, C.; Weaver, D.R.; Reppert, S.M. Direct association between mouse PERIOD and CKIepsilon is critical for a functioning circadian clock. Mol. Cell. Biol. 2004, 24, 584–594. [Google Scholar] [CrossRef]
- Cardone, L.; Hirayama, J.; Giordano, F.; Tamaru, T.; Palvimo, J.; Sassone-Corsi, P. Circadian clock control by SUMOylation of BMAL1. Science 2005, 309, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Yagita, K.; Tamanini, F.; Yasuda, M.; Hoeijmakers, J.H.; van der Horst, G.T.; Okamura, H. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. 2002, 21, 1301–1314. [Google Scholar] [CrossRef]
- Bjarnason, G.A.; Jordan, R. Circadian variation of cell proliferation and cell cycle protein expression in man: Clinical implications. Prog. Cell Cycle Res. 2000, 4, 193–206. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Antoch, M.P. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 2007, 17, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 302, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Grechez-Cassiau, A.; Rayet, B.; Guillaumond, F.; Teboul, M.; Delaunay, F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J. Biol. Chem. 2008, 283, 4535–4542. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef]
- Hua, H.; Wang, Y.; Wan, C.; Liu, Y.; Zhu, B.; Yang, C.; Wang, X.; Wang, Z.; Cornelissen-Guillaume, G.; Halberg, F. Circadian gene mPer2 overexpression induces cancer cell apoptosis. Cancer Sci. 2006, 97, 589–596. [Google Scholar] [CrossRef]
- Gotoh, T.; Vila-Caballer, M.; Santos, C.S.; Liu, J.; Yang, J.; Finkielstein, C.V. The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol. Biol. Cell 2014, 25, 3081–3093. [Google Scholar] [CrossRef]
- Zou, X.; Kim, D.W.; Gotoh, T.; Liu, J.; Kim, J.K.; Finkielstein, C.V. A Systems Biology Approach Identifies Hidden Regulatory Connections Between the Circadian and Cell-Cycle Checkpoints. Front. Physiol. 2020, 11, 327. [Google Scholar] [CrossRef]
- Miki, T.; Matsumoto, T.; Zhao, Z.; Lee, C.C. p53 regulates Period2 expression and the circadian clock. Nat. Commun. 2013, 4, 2444. [Google Scholar] [CrossRef]
- Unsal-Kacmaz, K.; Mullen, T.E.; Kaufmann, W.K.; Sancar, A. Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell. Biol. 2005, 25, 3109–3116. [Google Scholar] [CrossRef] [PubMed]
- Corellou, F.; Bisgrove, S.R.; Kropf, D.L.; Meijer, L.; Kloareg, B.; Bouget, F.Y. A S/M DNA replication checkpoint prevents nuclear and cytoplasmic events of cell division including centrosomal axis alignment and inhibits activation of cyclin-dependent kinase-like proteins in fucoid zygotes. Development 2000, 127, 1651–1660. [Google Scholar]
- Takai, H.; Tominaga, K.; Motoyama, N.; Minamishima, Y.A.; Nagahama, H.; Tsukiyama, T.; Ikeda, K.; Nakayama, K.; Nakanishi, M.; Nakayama, K. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000, 14, 1439–1447. [Google Scholar] [PubMed]
- Fu, L.; Kettner, N.M. The circadian clock in cancer development and therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar] [CrossRef]
- Haus, E.L.; Smolensky, M.H. Shift work and cancer risk: Potential mechanistic roles of circadian disruption, light at night, and sleep deprivation. Sleep Med. Rev. 2013, 17, 273–284. [Google Scholar] [CrossRef]
- Kelleher, F.C.; Rao, A.; Maguire, A. Circadian molecular clocks and cancer. Cancer Lett. 2014, 342, 9–18. [Google Scholar] [CrossRef]
- Lahti, T.; Merikanto, I.; Partonen, T. Circadian clock disruptions and the risk of cancer. Ann. Med. 2012, 44, 847–853. [Google Scholar] [CrossRef]
- West, A.C.; Bechtold, D.A. The cost of circadian desynchrony: Evidence, insights and open questions. Bioessays 2015, 37, 777–788. [Google Scholar] [CrossRef]
- Gauger, M.A.; Sancar, A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005, 65, 6828–6834. [Google Scholar] [CrossRef]
- Antoch, M.P.; Toshkov, I.; Kuropatwinski, K.K.; Jackson, M. Deficiency in PER proteins has no effect on the rate of spontaneous and radiation-induced carcinogenesis. Cell Cycle 2013, 12, 3673–3680. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Gombart, A.F.; Yi, W.S.; Koeffler, C.; Hofmann, W.K.; Koeffler, H.P. Transcription profiling of C/EBP targets identifies Per2 as a gene implicated in myeloid leukemia. Blood 2005, 106, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 2006, 22, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Coffelt, S.B.; Mao, L.; Yuan, L.; Cheng, Q.; Hill, S.M. Period-2: A tumor suppressor gene in breast cancer. J. Circadian Rhythms 2008, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Gery, S.; Dashti, A.; Yin, D.; Zhou, Y.; Gu, J.; Koeffler, H.P. A role for the clock gene per1 in prostate cancer. Cancer Res. 2009, 69, 7619–7625. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Choo, K.B.; Hou, M.F.; Yeh, K.T.; Kuo, S.J.; Chang, J.G. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis 2005, 26, 1241–1246. [Google Scholar] [CrossRef]
- Gery, S.; Komatsu, N.; Kawamata, N.; Miller, C.W.; Desmond, J.; Virk, R.K.; Marchevsky, A.; McKenna, R.; Taguchi, H.; Koeffler, H.P. Epigenetic silencing of the candidate tumor suppressor gene Per1 in non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 1399–1404. [Google Scholar] [CrossRef]
- Hsu, C.M.; Lin, S.F.; Lu, C.T.; Lin, P.M.; Yang, M.Y. Altered expression of circadian clock genes in head and neck squamous cell carcinoma. Tumour Biol. 2012, 33, 149–155. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Huang, W.; Reiter, R.J.; Ahmad, N. Melatonin resynchronizes dysregulated circadian rhythm circuitry in human prostate cancer cells. J. Pineal Res. 2010, 49, 60–68. [Google Scholar] [CrossRef]
- Lin, Y.M.; Chang, J.H.; Yeh, K.T.; Yang, M.Y.; Liu, T.C.; Lin, S.F.; Su, W.W.; Chang, J.G. Disturbance of circadian gene expression in hepatocellular carcinoma. Mol. Carcinog. 2008, 47, 925–933. [Google Scholar] [CrossRef]
- Litlekalsoy, J.; Rostad, K.; Kalland, K.H.; Hostmark, J.G.; Laerum, O.D. Expression of circadian clock genes and proteins in urothelial cancer is related to cancer-associated genes. BMC Cancer 2016, 16, 549. [Google Scholar] [CrossRef]
- Mazzoccoli, G.; Piepoli, A.; Carella, M.; Panza, A.; Pazienza, V.; Benegiamo, G.; Palumbo, O.; Ranieri, E. Altered expression of the clock gene machinery in kidney cancer patients. Biomed. Pharmacother. 2012, 66, 175–179. [Google Scholar] [CrossRef]
- Mostafaie, N.; Kallay, E.; Sauerzapf, E.; Bonner, E.; Kriwanek, S.; Cross, H.S.; Huber, K.R.; Krugluger, W. Correlated downregulation of estrogen receptor beta and the circadian clock gene Per1 in human colorectal cancer. Mol. Carcinog. 2009, 48, 642–647. [Google Scholar] [CrossRef]
- Pogue-Geile, K.L.; Lyons-Weiler, J.; Whitcomb, D.C. Molecular overlap of fly circadian rhythms and human pancreatic cancer. Cancer Lett. 2006, 243, 55–57. [Google Scholar] [CrossRef]
- Xia, H.C.; Niu, Z.F.; Ma, H.; Cao, S.Z.; Hao, S.C.; Liu, Z.T.; Wang, F. Deregulated expression of the Per1 and Per2 in human gliomas. Can. J. Neurol. Sci. 2010, 37, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Yang, Y.; Yang, K.; Zhao, D.; Tang, H.; Ran, X. Loss of the clock gene PER2 is associated with cancer development and altered expression of important tumor-related genes in oral cancer. Int. J. Oncol. 2018, 52, 279–287. [Google Scholar] [CrossRef]
- Yang, M.Y.; Chang, J.G.; Lin, P.M.; Tang, K.P.; Chen, Y.H.; Lin, H.Y.; Liu, T.C.; Hsiao, H.H.; Liu, Y.C.; Lin, S.F. Downregulation of circadian clock genes in chronic myeloid leukemia: Alternative methylation pattern of hPER3. Cancer Sci. 2006, 97, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Miyake, S.; Sumi, Y.; Yamaguchi, S.; Yasui, A.; Muijtjens, M.; Hoeijmakers, J.H.; van der Horst, G.T. Photic induction of mPer1 and mPer2 in cry-deficient mice lacking a biological clock. Science 1999, 286, 2531–2534. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Donehower, L.A.; Herron, A.J.; Moore, D.D.; Fu, L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS ONE 2010, 5, e10995. [Google Scholar] [CrossRef] [PubMed]
- Vitaterna, M.H.; Selby, C.P.; Todo, T.; Niwa, H.; Thompson, C.; Fruechte, E.M.; Hitomi, K.; Thresher, R.J.; Ishikawa, T.; Miyazaki, J.; et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl. Acad Sci. USA 1999, 96, 12114–12119. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, N.; Lee, J.H.; Gaddameedhi, S.; Sancar, A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc. Natl. Acad Sci. USA 2009, 106, 2841–2846. [Google Scholar] [CrossRef]
- Lee, J.H.; Sancar, A. Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc. Natl. Acad Sci. USA 2011, 108, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sancar, A. Circadian clock disruption improves the efficacy of chemotherapy through p73-mediated apoptosis. Proc. Natl. Acad Sci. USA 2011, 108, 10668–10672. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.L.; Papp, S.J.; Chan, A.B.; Henriksson, E.; Jordan, S.D.; Kriebs, A.; Nguyen, M.; Wallace, M.; Li, Z.; Metallo, C.M.; et al. CRY2 and FBXL3 Cooperatively Degrade c-MYC. Mol. Cell 2016, 64, 774–789. [Google Scholar] [CrossRef]
- Papp, S.J.; Huber, A.L.; Jordan, S.D.; Kriebs, A.; Nguyen, M.; Moresco, J.; Yates, J.R.; Lamia, K.A. DNA damage shifts circadian clock time via Hausp-dependent Cry1 stabilization. eLife 2015, 4. [Google Scholar] [CrossRef]
- Jung, C.H.; Kim, E.M.; Park, J.K.; Hwang, S.G.; Moon, S.K.; Kim, W.J.; Um, H.D. Bmal1 suppresses cancer cell invasion by blocking the phosphoinositide 3-kinase-Akt-MMP-2 signaling pathway. Oncol. Rep. 2013, 29, 2109–2113. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Li, X.; Li, B.; Li, Y.; Xia, K.; Yang, Y.; Aman, S.; Wang, M.; Wu, H. Circadian protein BMAL1 promotes breast cancer cell invasion and metastasis by up-regulating matrix metalloproteinase9 expression. Cancer Cell Int. 2019, 19, 182. [Google Scholar] [CrossRef]
- Zeng, Z.L.; Wu, M.W.; Sun, J.; Sun, Y.L.; Cai, Y.C.; Huang, Y.J.; Xian, L.J. Effects of the biological clock gene Bmal1 on tumour growth and anti-cancer drug activity. J. Biochem. 2010, 148, 319–326. [Google Scholar] [CrossRef]
- Jiang, W.; Zhao, S.; Jiang, X.; Zhang, E.; Hu, G.; Hu, B.; Zheng, P.; Xiao, J.; Lu, Z.; Lu, Y.; et al. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett. 2016, 371, 314–325. [Google Scholar] [CrossRef]
- Tang, Q.; Cheng, B.; Xie, M.; Chen, Y.; Zhao, J.; Zhou, X.; Chen, L. Circadian Clock Gene Bmal1 Inhibits Tumorigenesis and Increases Paclitaxel Sensitivity in Tongue Squamous Cell Carcinoma. Cancer Res. 2017, 77, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.L.; Luo, H.Y.; Yang, J.; Wu, W.J.; Chen, D.L.; Huang, P.; Xu, R.H. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin. Cancer Res. 2014, 20, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Vander Heiden, M.G.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Elshazley, M.; Sato, M.; Hase, T.; Yamashita, R.; Yoshida, K.; Toyokuni, S.; Ishiguro, F.; Osada, H.; Sekido, Y.; Yokoi, K.; et al. The circadian clock gene BMAL1 is a novel therapeutic target for malignant pleural mesothelioma. Int. J. Cancer 2012, 131, 2820–2831. [Google Scholar] [CrossRef] [PubMed]
- Puram, R.V.; Kowalczyk, M.S.; de Boer, C.G.; Schneider, R.K.; Miller, P.G.; McConkey, M.; Tothova, Z.; Tejero, H.; Heckl, D.; Jaras, M.; et al. Core Circadian Clock Genes Regulate Leukemia Stem Cells in AML. Cell 2016, 165, 303–316. [Google Scholar] [CrossRef]
- Korkmaz, T.; Aygenli, F.; Emisoglu, H.; Ozcelik, G.; Canturk, A.; Yilmaz, S.; Ozturk, N. Opposite Carcinogenic Effects of Circadian Clock Gene BMAL1. Sci. Rep. 2018, 8, 16023. [Google Scholar] [CrossRef]
- Hoffman, A.E.; Yi, C.H.; Zheng, T.; Stevens, R.G.; Leaderer, D.; Zhang, Y.; Holford, T.R.; Hansen, J.; Paulson, J.; Zhu, Y. CLOCK in breast tumorigenesis: Genetic, epigenetic, and transcriptional profiling analyses. Cancer Res. 2010, 70, 1459–1468. [Google Scholar] [CrossRef]
- Xiao, L.; Chang, A.K.; Zang, M.X.; Bi, H.; Li, S.; Wang, M.; Xing, X.; Wu, H. Induction of the CLOCK gene by E2-ERalpha signaling promotes the proliferation of breast cancer cells. PLoS ONE 2014, 9, e95878. [Google Scholar] [CrossRef]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar] [CrossRef]
- Sahar, S.; Sassone-Corsi, P. Circadian clock and breast cancer: A molecular link. Cell Cycle 2007, 6, 1329–1331. [Google Scholar] [CrossRef]
- Mormont, M.C.; Hecquet, B.; Bogdan, A.; Benavides, M.; Touitou, Y.; Levi, F. Non-invasive estimation of the circadian rhythm in serum cortisol in patients with ovarian or colorectal cancer. Int. J. Cancer 1998, 78, 421–424. [Google Scholar] [CrossRef]
- Mormont, M.C.; Levi, F. Circadian-system alterations during cancer processes: A review. Int. J. Cancer 1997, 70, 241–247. [Google Scholar] [CrossRef]
- Davis, S.; Mirick, D.K.; Stevens, R.G. Night shift work, light at night, and risk of breast cancer. J. Natl. Cancer Inst. 2001, 93, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Fritschi, L.; Erren, T.C.; Glass, D.C.; Girschik, J.; Thomson, A.K.; Saunders, C.; Boyle, T.; El-Zaemey, S.; Rogers, P.; Peters, S.; et al. The association between different night shiftwork factors and breast cancer: A case-control study. Br. J. Cancer 2013, 109, 2472–2480. [Google Scholar] [CrossRef]
- Ijaz, S.; Verbeek, J.; Seidler, A.; Lindbohm, M.L.; Ojajarvi, A.; Orsini, N.; Costa, G.; Neuvonen, K. Night-shift work and breast cancer--a systematic review and meta-analysis. Scand. J. Work Environ. Health 2013, 39, 431–447. [Google Scholar] [CrossRef]
- James, P.; Bertrand, K.A.; Hart, J.E.; Schernhammer, E.S.; Tamimi, R.M.; Laden, F. Outdoor Light at Night and Breast Cancer Incidence in the Nurses’ Health Study II. Environ. Health Perspect. 2017, 125, 087010. [Google Scholar] [CrossRef]
- Jia, Y.; Lu, Y.; Wu, K.; Lin, Q.; Shen, W.; Zhu, M.; Huang, S.; Chen, J. Does night work increase the risk of breast cancer? A systematic review and meta-analysis of epidemiological studies. Cancer Epidemiol. 2013, 37, 197–206. [Google Scholar] [CrossRef]
- Kamdar, B.B.; Tergas, A.I.; Mateen, F.J.; Bhayani, N.H.; Oh, J. Night-shift work and risk of breast cancer: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 138, 291–301. [Google Scholar] [CrossRef]
- Kojo, K.; Pukkala, E.; Auvinen, A. Breast cancer risk among Finnish cabin attendants: A nested case-control study. Occup. Environ. Med. 2005, 62, 488–493. [Google Scholar] [CrossRef]
- Lin, X.; Chen, W.; Wei, F.; Ying, M.; Wei, W.; Xie, X. Night-shift work increases morbidity of breast cancer and all-cause mortality: A meta-analysis of 16 prospective cohort studies. Sleep Med. 2015, 16, 1381–1387. [Google Scholar] [CrossRef]
- Megdal, S.P.; Kroenke, C.H.; Laden, F.; Pukkala, E.; Schernhammer, E.S. Night work and breast cancer risk: A systematic review and meta-analysis. Eur. J. Cancer 2005, 41, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Menegaux, F.; Truong, T.; Anger, A.; Cordina-Duverger, E.; Lamkarkach, F.; Arveux, P.; Kerbrat, P.; Fevotte, J.; Guenel, P. Night work and breast cancer: A population-based case-control study in France (the CECILE study). Int. J. Cancer 2013, 132, 924–931. [Google Scholar] [CrossRef]
- Pukkala, E.; Auvinen, A.; Wahlberg, G. Incidence of cancer among Finnish airline cabin attendants, 1967–1992. BMJ 1995, 311, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Schernhammer, E.S.; Kroenke, C.H.; Laden, F.; Hankinson, S.E. Night work and risk of breast cancer. Epidemiology 2006, 17, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Schernhammer, E.S.; Laden, F.; Speizer, F.E.; Willett, W.C.; Hunter, D.J.; Kawachi, I.; Colditz, G.A. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl. Cancer Inst. 2001, 93, 1563–1568. [Google Scholar] [CrossRef]
- Thompson, C.L.; Li, L. Association of sleep duration and breast cancer OncotypeDX recurrence score. Breast Cancer Res. Treat. 2012, 134, 1291–1295. [Google Scholar] [CrossRef]
- Travis, R.C.; Balkwill, A.; Fensom, G.K.; Appleby, P.N.; Reeves, G.K.; Wang, X.S.; Roddam, A.W.; Gathani, T.; Peto, R.; Green, J.; et al. Night Shift Work and Breast Cancer Incidence: Three Prospective Studies and Meta-analysis of Published Studies. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Wang, F.; Yeung, K.L.; Chan, W.C.; Kwok, C.C.; Leung, S.L.; Wu, C.; Chan, E.Y.; Yu, I.T.; Yang, X.R.; Tse, L.A. A meta-analysis on dose-response relationship between night shift work and the risk of breast cancer. Ann. Oncol. 2013, 24, 2724–2732. [Google Scholar] [CrossRef]
- Chu, L.W.; Zhu, Y.; Yu, K.; Zheng, T.; Yu, H.; Zhang, Y.; Sesterhenn, I.; Chokkalingam, A.P.; Danforth, K.N.; Shen, M.C.; et al. Variants in circadian genes and prostate cancer risk: A population-based study in China. Prostate Cancer Prostatic Dis. 2008, 11, 342–348. [Google Scholar] [CrossRef]
- Conlon, M.; Lightfoot, N.; Kreiger, N. Rotating shift work and risk of prostate cancer. Epidemiology 2007, 18, 182–183. [Google Scholar] [CrossRef]
- Flynn-Evans, E.E.; Mucci, L.; Stevens, R.G.; Lockley, S.W. Shiftwork and prostate-specific antigen in the National Health and Nutrition Examination Survey. J. Natl. Cancer Inst. 2013, 105, 1292–1297. [Google Scholar] [CrossRef]
- Kubo, T.; Oyama, I.; Nakamura, T.; Kunimoto, M.; Kadowaki, K.; Otomo, H.; Fujino, Y.; Fujimoto, N.; Matsumoto, T.; Matsuda, S. Industry-based retrospective cohort study of the risk of prostate cancer among rotating-shift workers. Int. J. Urol. 2011, 18, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Markt, S.C.; Grotta, A.; Nyren, O.; Adami, H.O.; Mucci, L.A.; Valdimarsdottir, U.A.; Stattin, P.; Bellocco, R.; Lagerros, Y.T. Insufficient Sleep and Risk of Prostate Cancer in a Large Swedish Cohort. Sleep 2015, 38, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Papantoniou, K.; Castano-Vinyals, G.; Espinosa, A.; Aragones, N.; Perez-Gomez, B.; Burgos, J.; Gomez-Acebo, I.; Llorca, J.; Peiro, R.; Jimenez-Moleon, J.; et al. Night shift work, chronotype and prostate cancer risk in the MCC-Spain case-control study. Int. J. Cancer 2015, 137, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Parent, M.E.; El-Zein, M.; Rousseau, M.C.; Pintos, J.; Siemiatycki, J. Night work and the risk of cancer among men. Am. J. Epidemiol. 2012, 176, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.; Yu, H.; Bai, Y.; Zheng, X.; Xie, L. Does night-shift work increase the risk of prostate cancer? A systematic review and meta-analysis. Onco Targets Ther. 2015, 8, 2817–2826. [Google Scholar] [CrossRef]
- Sigurdardottir, L.G.; Valdimarsdottir, U.A.; Fall, K.; Rider, J.R.; Lockley, S.W.; Schernhammer, E.; Mucci, L.A. Circadian disruption, sleep loss, and prostate cancer risk: A systematic review of epidemiologic studies. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1002–1011. [Google Scholar] [CrossRef]
- Wendeu-Foyet, M.G.; Koudou, Y.; Cenee, S.; Tretarre, B.; Rebillard, X.; Cancel-Tassin, G.; Cussenot, O.; Boland, A.; Bacq, D.; Deleuze, J.F.; et al. Circadian genes and risk of prostate cancer: Findings from the EPICAP study. Int. J. Cancer 2019, 145, 1745–1753. [Google Scholar] [CrossRef]
- Wendeu-Foyet, M.G.; Menegaux, F. Circadian Disruption and Prostate Cancer Risk: An Updated Review of Epidemiological Evidences. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 985–991. [Google Scholar] [CrossRef]
- Jiao, L.; Duan, Z.; Sangi-Haghpeykar, H.; Hale, L.; White, D.L.; El-Serag, H.B. Sleep duration and incidence of colorectal cancer in postmenopausal women. Br. J. Cancer 2013, 108, 213–221. [Google Scholar] [CrossRef]
- Thompson, C.L.; Larkin, E.K.; Patel, S.; Berger, N.A.; Redline, S.; Li, L. Short duration of sleep increases risk of colorectal adenoma. Cancer 2011, 117, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, A.N.; Hankinson, S.E.; Schernhammer, E.S. Night shift work and the risk of endometrial cancer. Cancer Res. 2007, 67, 10618–10622. [Google Scholar] [CrossRef] [PubMed]
- Lahti, T.A.; Partonen, T.; Kyyronen, P.; Kauppinen, T.; Pukkala, E. Night-time work predisposes to non-Hodgkin lymphoma. Int. J. Cancer 2008, 123, 2148–2151. [Google Scholar] [CrossRef] [PubMed]
- Mormont, M.C.; Waterhouse, J.; Bleuzen, P.; Giacchetti, S.; Jami, A.; Bogdan, A.; Lellouch, J.; Misset, J.L.; Touitou, Y.; Levi, F. Marked 24-h rest/activity rhythms are associated with better quality of life, better response, and longer survival in patients with metastatic colorectal cancer and good performance status. Clin. Cancer Res. 2000, 6, 3038–3045. [Google Scholar]
- Baan, R.; Grosse, Y.; Straif, K.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part F: Chemical agents and related occupations. Lancet Oncol. 2009, 10, 1143–1144. [Google Scholar] [CrossRef]
- Straif, K.; Baan, R.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Benbrahim-Tallaa, L.; Cogliano, V.; Group WHOIAFRoCM. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007, 8, 1065–1066. [Google Scholar] [CrossRef]
- Costa, G.; Haus, E.; Stevens, R. Shift work and cancer-considerations on rationale, mechanisms, and epidemiology. Scand. J. Work Environ. Health 2010, 36, 163–179. [Google Scholar] [CrossRef]
- Fritschi, L.; Glass, D.C.; Heyworth, J.S.; Aronson, K.; Girschik, J.; Boyle, T.; Grundy, A.; Erren, T.C. Hypotheses for mechanisms linking shiftwork and cancer. Med. Hypotheses 2011, 77, 430–436. [Google Scholar] [CrossRef]
- Dierickx, P.; Van Laake, L.W.; Geijsen, N. Circadian clocks: From stem cells to tissue homeostasis and regeneration. EMBO Rep. 2018, 19, 18–28. [Google Scholar] [CrossRef]
- Dierickx, P.; Vermunt, M.W.; Muraro, M.J.; Creyghton, M.P.; Doevendans, P.A.; van Oudenaarden, A.; Geijsen, N.; Van Laake, L.W. Circadian networks in human embryonic stem cell-derived cardiomyocytes. EMBO Rep. 2017, 18, 1199–1212. [Google Scholar] [CrossRef]
- Paatela, E.; Munson, D.; Kikyo, N. Circadian Regulation in Tissue Regeneration. Int. J. Mol. Sci. 2019, 20, 2263. [Google Scholar] [CrossRef] [PubMed]
- Weger, M.; Diotel, N.; Dorsemans, A.C.; Dickmeis, T.; Weger, B.D. Stem cells and the circadian clock. Dev. Biol. 2017, 431, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Umemura, Y.; Koike, N.; Ohashi, M.; Tsuchiya, Y.; Meng, Q.J.; Minami, Y.; Hara, M.; Hisatomi, M.; Yagita, K. Involvement of posttranscriptional regulation of Clock in the emergence of circadian clock oscillation during mouse development. Proc. Natl. Acad Sci. USA 2017, 114, E7479–E7488. [Google Scholar] [CrossRef] [PubMed]
- Yagita, K.; Horie, K.; Koinuma, S.; Nakamura, W.; Yamanaka, I.; Urasaki, A.; Shigeyoshi, Y.; Kawakami, K.; Shimada, S.; Takeda, J.; et al. Development of the circadian oscillator during differentiation of mouse embryonic stem cells in vitro. Proc. Natl. Acad Sci. USA 2010, 107, 3846–3851. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef]
- Zheng, B.; Larkin, D.W.; Albrecht, U.; Sun, Z.S.; Sage, M.; Eichele, G.; Lee, C.C.; Bradley, A. The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature 1999, 400, 169–173. [Google Scholar] [CrossRef]
- Brown, S.A. Circadian clock-mediated control of stem cell division and differentiation: Beyond night and day. Development 2014, 141, 3105–3111. [Google Scholar] [CrossRef]
- Plikus, M.V.; Chuong, C.M. Complex hair cycle domain patterns and regenerative hair waves in living rodents. J. Investig. Dermatol. 2008, 128, 1071–1080. [Google Scholar] [CrossRef]
- Gaddameedhi, S.; Selby, C.P.; Kaufmann, W.K.; Smart, R.C.; Sancar, A. Control of skin cancer by the circadian rhythm. Proc. Natl. Acad Sci. USA 2011, 108, 18790–18795. [Google Scholar] [CrossRef]
- Janich, P.; Toufighi, K.; Solanas, G.; Luis, N.M.; Minkwitz, S.; Serrano, L.; Lehner, B.; Benitah, S.A. Human epidermal stem cell function is regulated by circadian oscillations. Cell Stem Cell 2013, 13, 745–753. [Google Scholar] [CrossRef]
- Lin, K.K.; Kumar, V.; Geyfman, M.; Chudova, D.; Ihler, A.T.; Smyth, P.; Paus, R.; Takahashi, J.S.; Andersen, B. Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet. 2009, 5, e1000573. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Van Spyk, E.N.; Pham, K.; Geyfman, M.; Kumar, V.; Takahashi, J.S.; Andersen, B. The circadian clock in skin: Implications for adult stem cells, tissue regeneration, cancer, aging, and immunity. J. Biol. Rhythms 2015, 30, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, N.P.; Seinkmane, E.; Putker, M.; Feeney, K.A.; Krogager, T.P.; Chesham, J.E.; Bray, L.K.; Thomas, J.M.; Dunn, K.; Blaikley, J.; et al. Circadian actin dynamics drive rhythmic fibroblast mobilization during wound healing. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Durgan, D.J.; Young, M.E. The cardiomyocyte circadian clock: Emerging roles in health and disease. Circ. Res. 2010, 106, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Yao, H.; Sellix, M.T.; Rahman, I. Circadian molecular clock in lung pathophysiology. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1056–L1075. [Google Scholar] [CrossRef]
- Tsinkalovsky, O.; Filipski, E.; Rosenlund, B.; Sothern, R.B.; Eiken, H.G.; Wu, M.W.; Claustrat, B.; Bayer, J.; Levi, F.; Laerum, O.D. Circadian expression of clock genes in purified hematopoietic stem cells is developmentally regulated in mouse bone marrow. Exp. Hematol. 2006, 34, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Tsinkalovsky, O.; Rosenlund, B.; Laerum, O.D.; Eiken, H.G. Clock gene expression in purified mouse hematopoietic stem cells. Exp. Hematol. 2005, 33, 100–107. [Google Scholar] [CrossRef]
- Karpowicz, P.; Zhang, Y.; Hogenesch, J.B.; Emery, P.; Perrimon, N. The circadian clock gates the intestinal stem cell regenerative state. Cell Rep. 2013, 3, 996–1004. [Google Scholar] [CrossRef]
- Konturek, P.C.; Brzozowski, T.; Konturek, S.J. Stress and the gut: Pathophysiology, clinical consequences, diagnostic approach and treatment options. J. Physiol. Pharmacol. 2011, 62, 591–599. [Google Scholar]
- Pagel, R.; Bar, F.; Schroder, T.; Sunderhauf, A.; Kunstner, A.; Ibrahim, S.M.; Autenrieth, S.E.; Kalies, K.; Konig, P.; Tsang, A.H.; et al. Circadian rhythm disruption impairs tissue homeostasis and exacerbates chronic inflammation in the intestine. FASEB J. 2017, 31, 4707–4719. [Google Scholar] [CrossRef]
- Stokes, K.; Cooke, A.; Chang, H.; Weaver, D.R.; Breault, D.T.; Karpowicz, P. The Circadian Clock Gene BMAL1 Coordinates Intestinal Regeneration. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 95–114. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hou, L.; Xia, H.; Li, H.; Fan, H.; Jia, X.; Niu, Z. PER2 inhibits proliferation and stemness of glioma stem cells via the Wnt/betacatenin signaling pathway. Oncol. Rep. 2020, 44, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sun, H.; Zhang, S.; Yang, X.; Zhang, G.; Su, T. Overexpression of PER3 Inhibits Self-Renewal Capability and Chemoresistance of Colorectal Cancer Stem-Like Cells via Inhibition of Notch and beta-Catenin Signaling. Oncol. Res. 2017, 25, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Ogino, T.; Hara, Y.; Tanaka, T.; Koyanagi, S.; Ohdo, S. Optimized Dosing Schedule Based on Circadian Dynamics of Mouse Breast Cancer Stem Cells Improves the Antitumor Effects of Aldehyde Dehydrogenase Inhibitor. Cancer Res. 2018, 78, 3698–3708. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef]
- DeLellis, R.A. Pathology and genetics of thyroid carcinoma. J. Surg. Oncol. 2006, 94, 662–669. [Google Scholar] [CrossRef]
- Jin, S.; Borkhuu, O.; Bao, W.; Yang, Y.T. Signaling Pathways in Thyroid Cancer and Their Therapeutic Implications. J. Clin. Med. Res. 2016, 8, 284–296. [Google Scholar] [CrossRef]
- Nikiforov, Y.E. Thyroid carcinoma: Molecular pathways and therapeutic targets. Mod. Pathol. 2008, 21 (Suppl. 2), S37–S43. [Google Scholar] [CrossRef]
- De Felice, M.; Postiglione, M.P.; Di Lauro, R. Minireview: Thyrotropin receptor signaling in development and differentiation of the thyroid gland: Insights from mouse models and human diseases. Endocrinology 2004, 145, 4062–4067. [Google Scholar] [CrossRef]
- Franco, A.T.; Malaguarnera, R.; Refetoff, S.; Liao, X.H.; Lundsmith, E.; Kimura, S.; Pritchard, C.; Marais, R.; Davies, T.F.; Weinstein, L.S.; et al. Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proc. Natl. Acad Sci. USA 2011, 108, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Van Keymeulen, A.; Golstein, J.; Fusco, A.; Dumont, J.E.; Roger, P.P. Regulation of thyroid cell proliferation by TSH and other factors: A critical evaluation of in vitro models. Endocr. Rev. 2001, 22, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Chen, K.Y.; Kim, T.Y.; Dominguez, J.M.; Voza, F.; Ouyang, B.; Vundavalli, S.K.; Knauf, J.A.; Fagin, J.A. Switch in signaling control of mTORC1 activity after oncoprotein expression in thyroid cancer cell lines. J. Clin. Endocrinol. Metab. 2014, 99, E1976–E1987. [Google Scholar] [CrossRef] [PubMed]
- Nozhat, Z.; Hedayati, M. PI3K/AKT Pathway and Its Mediators in Thyroid Carcinomas. Mol. Diagn. Ther. 2016, 20, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Gimm, O.; Attie-Bitach, T.; Lees, J.A.; Vekemans, M.; Eng, C. Expression of the PTEN tumour suppressor protein during human development. Hum. Mol. Genet. 2000, 9, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Morcavallo, A.; Belfiore, A. The insulin and igf-I pathway in endocrine glands carcinogenesis. J. Oncol. 2012, 2012, 635614. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Belfiore, A. The insulin receptor: A new target for cancer therapy. Front. Endocrinol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Vella, V.; Pandini, G.; Sciacca, L.; Mineo, R.; Vigneri, R.; Pezzino, V.; Belfiore, A. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J. Clin. Endocrinol. Metab. 2002, 87, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Belfiore, A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front. Endocrinol. 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Morcavallo, A.; Giuliano, S.; Belfiore, A. Thyroid cancer development and progression: Emerging role of cancer stem cells. Minerva Endocrinol. 2012, 37, 103–115. [Google Scholar] [PubMed]
- Belfiore, A.; Gangemi, P.; Costantino, A.; Russo, G.; Santonocito, G.M.; Ippolito, O.; Di Renzo, M.F.; Comoglio, P.; Fiumara, A.; Vigneri, R. Negative/low expression of the Met/hepatocyte growth factor receptor identifies papillary thyroid carcinomas with high risk of distant metastases. J. Clin. Endocrinol. Metab. 1997, 82, 2322–2328. [Google Scholar] [CrossRef]
- Di Renzo, M.F.; Olivero, M.; Ferro, S.; Prat, M.; Bongarzone, I.; Pilotti, S.; Belfiore, A.; Costantino, A.; Vigneri, R.; Pierotti, M.A.; et al. Overexpression of the c-MET/HGF receptor gene in human thyroid carcinomas. Oncogene 1992, 7, 2549–2553. [Google Scholar]
- Mineo, R.; Costantino, A.; Frasca, F.; Sciacca, L.; Russo, S.; Vigneri, R.; Belfiore, A. Activation of the hepatocyte growth factor (HGF)-Met system in papillary thyroid cancer: Biological effects of HGF in thyroid cancer cells depend on Met expression levels. Endocrinology 2004, 145, 4355–4365. [Google Scholar] [CrossRef]
- Ruco, L.; Scarpino, S. The Pathogenetic Role of the HGF/c-Met System in Papillary Carcinoma of the Thyroid. Biomedicines 2014, 2, 263–274. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Nicolosi, M.L.; Sacco, A.; Morcavallo, A.; Vella, V.; Voci, C.; Spatuzza, M.; Xu, S.Q.; Iozzo, R.V.; Vigneri, R.; et al. Novel cross talk between IGF-IR and DDR1 regulates IGF-IR trafficking, signaling and biological responses. Oncotarget 2015, 6, 16084–16105. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Vella, V.; Pellegriti, G.; Belfiore, A. Editorial: Clinical and Molecular Epidemiology of Thyroid Cancer of Follicular Origin. Front. Endocrinol. 2018, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R. The Emerging Role of Insulin Receptor Isoforms in Thyroid Cancer: Clinical Implications and New Perspectives. Int. J. Mol. Sci. 2018, 19, 3814. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R.; Nicolosi, M.L.; Morrione, A.; Belfiore, A. Insulin/IGF signaling and discoidin domain receptors: An emerging functional connection. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118522. [Google Scholar] [CrossRef]
- Vella, V.; Nicolosi, M.L.; Cantafio, P.; Massimino, M.; Lappano, R.; Vigneri, P.; Ciuni, R.; Gangemi, P.; Morrione, A.; Malaguarnera, R.; et al. DDR1 regulates thyroid cancer cell differentiation via IGF-2/IR-A autocrine signaling loop. Endocr. Relat. Cancer 2019, 26, 197–214. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Vella, V.; Aloisi, A.; Mandarino, A.; Mazzon, E.; Vigneri, R.; Vigneri, P. p73 tumor-suppressor activity is impaired in human thyroid cancer. Cancer Res. 2003, 63, 5829–5837. [Google Scholar]
- Kunstman, J.W.; Juhlin, C.C.; Goh, G.; Brown, T.C.; Stenman, A.; Healy, J.M.; Rubinstein, J.C.; Choi, M.; Kiss, N.; Nelson-Williams, C.; et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum. Mol. Genet. 2015, 24, 2318–2329. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Mandarino, A.; Mazzon, E.; Vella, V.; Gangemi, P.; Vancheri, C.; Vigneri, P.; Aloisi, A.; Vigneri, R.; Frasca, F. The p53-homologue p63 may promote thyroid cancer progression. Endocr. Relat. Cancer 2005, 12, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Vella, V.; Pandini, G.; Sanfilippo, M.; Pezzino, V.; Vigneri, R.; Frasca, F. TAp73 alpha increases p53 tumor suppressor activity in thyroid cancer cells via the inhibition of Mdm2-mediated degradation. Mol. Cancer Res. 2008, 6, 64–77. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Vella, V.; Vigneri, R.; Frasca, F. p53 family proteins in thyroid cancer. Endocr. Relat. Cancer 2007, 14, 43–60. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Sykorova, V.; Dvorakova, S.; Vcelak, J.; Vaclavikova, E.; Halkova, T.; Kodetova, D.; Lastuvka, P.; Betka, J.; Vlcek, P.; Reboun, M.; et al. Search for new genetic biomarkers in poorly differentiated and anaplastic thyroid carcinomas using next generation sequencing. Anticancer Res. 2015, 35, 2029–2036. [Google Scholar] [PubMed]
- Xu, B.; Ghossein, R. Genomic Landscape of poorly Differentiated and Anaplastic Thyroid Carcinoma. Endocr. Pathol. 2016, 27, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, Y.; Shimamura, M.; Mitsutake, N. Cancer Stem Cells in the Thyroid. Front. Endocrinol. 2016, 7, 20. [Google Scholar] [CrossRef]
- Zane, M.; Scavo, E.; Catalano, V.; Bonanno, M.; Todaro, M.; De Maria, R.; Stassi, G. Normal vs. cancer thyroid stem cells: The road to transformation. Oncogene 2016, 35, 805–815. [Google Scholar] [CrossRef]
- Takano, T. Fetal cell carcinogenesis of the thyroid: A hypothesis for better understanding of gene expression profile and genomic alternation in thyroid carcinoma. Endocr. J. 2004, 51, 509–515. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Frasca, F.; Garozzo, A.; Giani, F.; Pandini, G.; Vella, V.; Vigneri, R.; Belfiore, A. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J. Clin. Endocrinol. Metab. 2011, 96, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar] [PubMed]
- Lowry, W.E.; Richter, L. Signaling in adult stem cells. Front. Biosci. 2007, 12, 3911–3927. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, K.; Matsuse, M.; Nakazawa, Y.; Ogi, T.; Suzuki, K.; Saenko, V.; Xu, S.; Umezawa, K.; Yamashita, S.; Tsukamoto, K.; et al. JAK/STAT3 and NF-kappaB Signaling Pathways Regulate Cancer Stem-Cell Properties in Anaplastic Thyroid Cancer Cells. Thyroid 2019, 29, 674–682. [Google Scholar] [CrossRef]
- Todaro, M.; Iovino, F.; Eterno, V.; Cammareri, P.; Gambara, G.; Espina, V.; Gulotta, G.; Dieli, F.; Giordano, S.; De Maria, R.; et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 2010, 70, 8874–8885. [Google Scholar] [CrossRef]
- Forte, S.; La Rosa, C.; Pecce, V.; Rosignolo, F.; Memeo, L. The role of microRNAs in thyroid carcinomas. Anticancer Res. 2015, 35, 2037–2047. [Google Scholar]
- He, H.; Olesnanik, K.; Nagy, R.; Liyanarachchi, S.; Prasad, M.L.; Stratakis, C.A.; Kloos, R.T.; de la Chapelle, A. Allelic variation in gene expression in thyroid tissue. Thyroid 2005, 15, 660–667. [Google Scholar] [CrossRef]
- Pallante, P.; Visone, R.; Ferracin, M.; Ferraro, A.; Berlingieri, M.T.; Troncone, G.; Chiappetta, G.; Liu, C.G.; Santoro, M.; Negrini, M.; et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr. Relat. Cancer 2006, 13, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Chen, Y.; Gong, Y.; Dong, T.; Zhang, B.; Gao, W. miR-148a inhibits self-renewal of thyroid cancer stem cells via repressing INO80 expression. Oncol. Rep. 2016, 36, 3387–3396. [Google Scholar] [CrossRef] [PubMed]
- Haghpanah, V.; Fallah, P.; Tavakoli, R.; Naderi, M.; Samimi, H.; Soleimani, M.; Larijani, B. Antisense-miR-21 enhances differentiation/apoptosis and reduces cancer stemness state on anaplastic thyroid cancer. Tumour Biol. 2016, 37, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chai, H.F.; Peng, F.; Meng, Y.T.; Zhang, L.Z.; Zhang, L.; Zou, H.; Liang, Q.L.; Li, M.M.; Mao, K.G.; et al. Estrogen receptor beta upregulated by lncRNA-H19 to promote cancer stem-like properties in papillary thyroid carcinoma. Cell Death Dis. 2018, 9, 1120. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, P.X.; Cheng, J.W.; Sun, Y.F.; Hu, B.; Guo, W.; Zhou, K.Q.; Yin, Y.; Li, Y.C.; Wang, J.; et al. Chemotherapeutic perfusion of portal vein after tumor thrombectomy and hepatectomy benefits patients with advanced hepatocellular carcinoma: A propensity score-matched survival analysis. Cancer Med. 2019, 8, 6933–6944. [Google Scholar] [CrossRef] [PubMed]
- Gamble, K.L.; Berry, R.; Frank, S.J.; Young, M.E. Circadian clock control of endocrine factors. Nat. Rev. Endocrinol. 2014, 10, 466–475. [Google Scholar] [CrossRef]
- Weeke, J.; Gundersen, H.J. Circadian and 30 minutes variations in serum TSH and thyroid hormones in normal subjects. Acta Endocrinol. 1978, 89, 659–672. [Google Scholar] [CrossRef]
- Fahrenkrug, J.; Georg, B.; Hannibal, J.; Jorgensen, H.L. Hypophysectomy abolishes rhythms in rat thyroid hormones but not in the thyroid clock. J. Endocrinol. 2017, 233, 209–216. [Google Scholar] [CrossRef]
- Gronfier, C.; Brandenberger, G. Ultradian rhythms in pituitary and adrenal hormones: Their relations to sleep. Sleep Med. Rev. 1998, 2, 17–29. [Google Scholar] [CrossRef]
- Magrini, A.; Pietroiusti, A.; Coppeta, L.; Babbucci, A.; Barnaba, E.; Papadia, C.; Iannaccone, U.; Boscolo, P.; Bergamaschi, E.; Bergamaschi, A. Shift work and autoimmune thyroid disorders. Int. J. Immunopathol. Pharmacol. 2006, 19, 31–36. [Google Scholar]
- Amir, S.; Robinson, B. Thyroidectomy alters the daily pattern of expression of the clock protein, PER2, in the oval nucleus of the bed nucleus of the stria terminalis and central nucleus of the amygdala in rats. Neurosci. Lett. 2006, 407, 254–257. [Google Scholar] [CrossRef]
- Bargi-Souza, P.; Peliciari-Garcia, R.A.; Nunes, M.T. Disruption of the Pituitary Circadian Clock Induced by Hypothyroidism and Hyperthyroidism: Consequences on Daily Pituitary Hormone Expression Profiles. Thyroid 2019, 29, 502–512. [Google Scholar] [CrossRef]
- Peliciari-Garcia, R.A.; Bargi-Souza, P.; Young, M.E.; Nunes, M.T. Repercussions of hypo and hyperthyroidism on the heart circadian clock. Chronobiol. Int. 2018, 35, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Peliciari-Garcia, R.A.; Previde, R.M.; Nunes, M.T.; Young, M.E. Interrelationship between 3,5,3 -triiodothyronine and the circadian clock in the rodent heart. Chronobiol. Int. 2016, 33, 1444–1454. [Google Scholar] [CrossRef]
- Hirschfeld, U.; Moreno-Reyes, R.; Akseki, E.; L’Hermite-Baleriaux, M.; Leproult, R.; Copinschi, G.; Van Cauter, E. Progressive elevation of plasma thyrotropin during adaptation to simulated jet lag: Effects of treatment with bright light or zolpidem. J. Clin. Endocrinol. Metab. 1996, 81, 3270–3277. [Google Scholar] [CrossRef]
- Ledda, C.; Cina, D.; Matera, S.; Mucci, N.; Bracci, M.; Rapisarda, V. High HOMA-IR Index in Healthcare Shift Workers. Medicina 2019, 55, 186. [Google Scholar] [CrossRef]
- Liu, G.S.; Cook, A.; Richardson, M.; Vail, D.; Holsinger, F.C.; Oakley-Girvan, I. Thyroid cancer risk in airline cockpit and cabin crew: A meta-analysis. Cancers Head Neck 2018, 3, 7. [Google Scholar] [CrossRef]
- Pinkerton, L.E.; Hein, M.J.; Anderson, J.L.; Christianson, A.; Little, M.P.; Sigurdson, A.J.; Schubauer-Berigan, M.K. Melanoma, thyroid cancer, and gynecologic cancers in a cohort of female flight attendants. Am. J. Ind. Med. 2018, 61, 572–581. [Google Scholar] [CrossRef]
- Luo, J.; Sands, M.; Wactawski-Wende, J.; Song, Y.; Margolis, K.L. Sleep disturbance and incidence of thyroid cancer in postmenopausal women the Women’s Health Initiative. Am. J. Epidemiol. 2013, 177, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Morales-Santana, S.; Morell, S.; Leon, J.; Carazo-Gallego, A.; Jimenez-Lopez, J.C.; Morell, M. An Overview of the Polymorphisms of Circadian Genes Associated with Endocrine Cancer. Front. Endocrinol. 2019, 10, 104. [Google Scholar] [CrossRef]
- Gallo, C.; Fragliasso, V.; Donati, B.; Torricelli, F.; Tameni, A.; Piana, S.; Ciarrocchi, A. The bHLH transcription factor DEC1 promotes thyroid cancer aggressiveness by the interplay with NOTCH1. Cell Death Dis. 2018, 9, 871. [Google Scholar] [CrossRef] [PubMed]
- Mond, M.; Alexiadis, M.; Eriksson, N.; Davis, M.J.; Muscat, G.E.; Fuller, P.J.; Gilfillan, C. Nuclear receptor expression in human differentiated thyroid tumors. Thyroid 2014, 24, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, L.; Grosclaude, P. [Epidemiology of thyroid carcinoma over the world]. Ann. Endocrinol. 2011, 72, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Copinschi, G.; Spiegel, K.; Leproult, R.; Van Cauter, E. Pathophysiology of human circadian rhythms. Novartis Found. Symp. 2000, 227, 143–157; discussion 157–162. [Google Scholar] [CrossRef] [PubMed]
- Maury, E.; Ramsey, K.M.; Bass, J. Circadian rhythms and metabolic syndrome: From experimental genetics to human disease. Circ. Res. 2010, 106, 447–462. [Google Scholar] [CrossRef]
- Ledda, C.; Rapisarda, V. Occupational and Environmental Carcinogenesis. Cancers 2020, 12, 2547. [Google Scholar] [CrossRef]
- Relogio, A.; Thomas, P.; Medina-Perez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schafer, R.; et al. Ras-mediated deregulation of the circadian clock in cancer. PLoS Genet. 2014, 10, e1004338. [Google Scholar] [CrossRef]
- Kettner, N.M.; Mayo, S.A.; Hua, J.; Lee, C.; Moore, D.D.; Fu, L. Circadian Dysfunction Induces Leptin Resistance in Mice. Cell Metab. 2015, 22, 448–459. [Google Scholar] [CrossRef]
- Pan, X.; Bradfield, C.A.; Hussain, M.M. Global and hepatocyte-specific ablation of Bmal1 induces hyperlipidaemia and enhances atherosclerosis. Nat. Commun. 2016, 7, 13011. [Google Scholar] [CrossRef]
- Shi, S.Q.; Ansari, T.S.; McGuinness, O.P.; Wasserman, D.H.; Johnson, C.H. Circadian disruption leads to insulin resistance and obesity. Curr. Biol. 2013, 23, 372–381. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Function/Signaling Pathway |
|---|---|---|
| AKT or PKB (Protein kinase B) | Protein kinase B (PKB) | Survival, proliferation, apoptosis resistance. PI3K/AKT signaling pathway |
| ATM (ataxia telangiectasia) | ATM | DNA damage response, cell cycle, apoptosis, mitochondrial homeostasis |
| ATR (ataxia telangiectasia and Rad-3 related protein) | ATR | DNA damage response, cell cycle. PI3K/AKT signaling pathway |
| BMAL1 (aryl hydrocarbon receptor nuclear translocator like) | BMAL1 | Circadian clock, exercise-induced circadian regulation, melatonin metabolism and effects, bone metabolism, energetic metabolism, cell stress |
| BRAF (B-Raf proto-oncogene, serine/threonine kinase) | BRAF | Oncogene. Proliferation, differentiation. MAPK/ERK signaling pathway |
| c-MYC | C-MYC | Proto-oncogene, transcription factor. Cell growth, apoptosis, differentiation, stem cell self-renewal. |
| CK1 (casein kinase 1) | CK1α-β-γ-δ-ε casein kinase 1α-β-γ-δ-ε) | Tumor suppressor. Circadian clock, metabolism, DNA damage, cellular stress, cell cycle, cytoskeleton associated functions. Developmental pathways |
| CLOCK (clock circadian regulator) | CLOCK | Circadian clock, exercise-induced circadian regulation, melatonin metabolism and effects. |
| CRYs (cryptochrome circadian regulators) | CRY1-2 | Tumor suppressor. Circadian clock. |
| DEC1-2 (differentially expressed in chondrocytes 1-2) | DEC1-2 | Tumor suppressor. Circadian clock. |
| INO80 | Chormatin-remodeling ATPase INO80 | Cell cycle, cell division, DNA damage, DNA recombination, DNA repair, mitosis, chromatin remodeling. |
| MEN1 | Menin | Transcriptional regulator. Telomerase repressor. Cell proliferation, DNA repair. TGFB1 and NFkB signaling |
| MET | Proto-oncogene c-Met | Proliferation, scattering, morphogenesis, survival, differentiation, angiogenesis. RAS/ERK, PI3K/AKT, PLC-γ/PKC signaling |
| NF1 (neurofibromatosis Type 1 Protein) | Neurofibromin 1 | Tumor suppressor. Cell growth and division. Ras inhibition. Circadian clock. |
| NPAS2 (neuronal PAS domain protein 2) | NPAS2 | Tumor suppressor. DNA damage response. Negative regulator of cell death. Circadian clock. Central nervous system development. Metabolism. |
| P53 | P53 | Tumor suppressor. Response to DNA damage. Cell cycle arrest. Apoptosis. Aging. Gene expression |
| PI3KCA (phosphatidylinositol-4,5-bisphosphate 3-kinase 110 kDa catalytic subunit alpha) | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha isoform | Catalytic activity (performs the action of PI3K). Proliferation, survival, migration. PI3K/AKT/mTOR signaling pathway. |
| PER1-2-3 (Period circadian regulator 1-2-3) | PER1-2-3 | Tumor suppressor. Circadian clock. Exercise-induced circadian regulation. Melatonin metabolism and effects. Chromatin DNA binding |
| PTEN (phosphatase and tensin homolog) | PTEN | Tumor suppressor. AKT/PKB signaling pathway |
| RAS | RAS | Oncogene. Cell growth, differentiation, survival, Cell adhesion, apoptosis, migration. MAPK/ERK and PI3K/AKT/mTOR pathway |
| RB1 (RB transcriptional corepressor 1) | Retinoblastoma associated protein RB1 | Tumor suppressor. Cell cycle. Chromatin remodeling. Cell differentiation, cell growth |
| REV-ERB or NR1D1 (nuclear receptor subfamily 1 group D member 1) | NR1D1 | Tumor suppressor. Circadian clock. Mitochondrial biogenesis. Nuclear Receptor transcription pathway |
| RET (RET proto-oncogene) | RET | Proto-oncogene. Protein tyrosine kinase activity. MAPK signaling. |
| RORα (RAR related orphan receptor A) | Nuclear receptor ROR-alpha | Tumor suppressor. Circadian clock. Metabolism. Transcription factor activity. |
| SWI/SNF (SMARCC1) | SWI/SNF complex subunit SMARCC1 | Chromatin remodeling. Transcription regulator |
| TERT (telomerase reverse transcriptase) | Telomerase reverse transcriptase | Chromosome replication. Telomerase activity. Transcription regulator |
| TIM (timeless circadian regulator) | Protein timeless homolog, hTIM | Tumor suppressor. Circadian clock. DNA replication, replication fork stability |
| WEE1 | WEE1-like protein kinase | Cell division, cell cycle, microtubule cytoskeleton organization |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malaguarnera, R.; Ledda, C.; Filippello, A.; Frasca, F.; Francavilla, V.C.; Ramaci, T.; Parisi, M.C.; Rapisarda, V.; Piro, S. Thyroid Cancer and Circadian Clock Disruption. Cancers 2020, 12, 3109. https://doi.org/10.3390/cancers12113109
Malaguarnera R, Ledda C, Filippello A, Frasca F, Francavilla VC, Ramaci T, Parisi MC, Rapisarda V, Piro S. Thyroid Cancer and Circadian Clock Disruption. Cancers. 2020; 12(11):3109. https://doi.org/10.3390/cancers12113109
Chicago/Turabian StyleMalaguarnera, Roberta, Caterina Ledda, Agnese Filippello, Francesco Frasca, Vincenzo Cristian Francavilla, Tiziana Ramaci, Maria Chiara Parisi, Venerando Rapisarda, and Salvatore Piro. 2020. "Thyroid Cancer and Circadian Clock Disruption" Cancers 12, no. 11: 3109. https://doi.org/10.3390/cancers12113109
APA StyleMalaguarnera, R., Ledda, C., Filippello, A., Frasca, F., Francavilla, V. C., Ramaci, T., Parisi, M. C., Rapisarda, V., & Piro, S. (2020). Thyroid Cancer and Circadian Clock Disruption. Cancers, 12(11), 3109. https://doi.org/10.3390/cancers12113109