Therapeutic Potential of PI3K/AKT/mTOR Pathway in Gastrointestinal Stromal Tumors: Rationale and Progress

Simple Summary
Abstract
1. Introduction
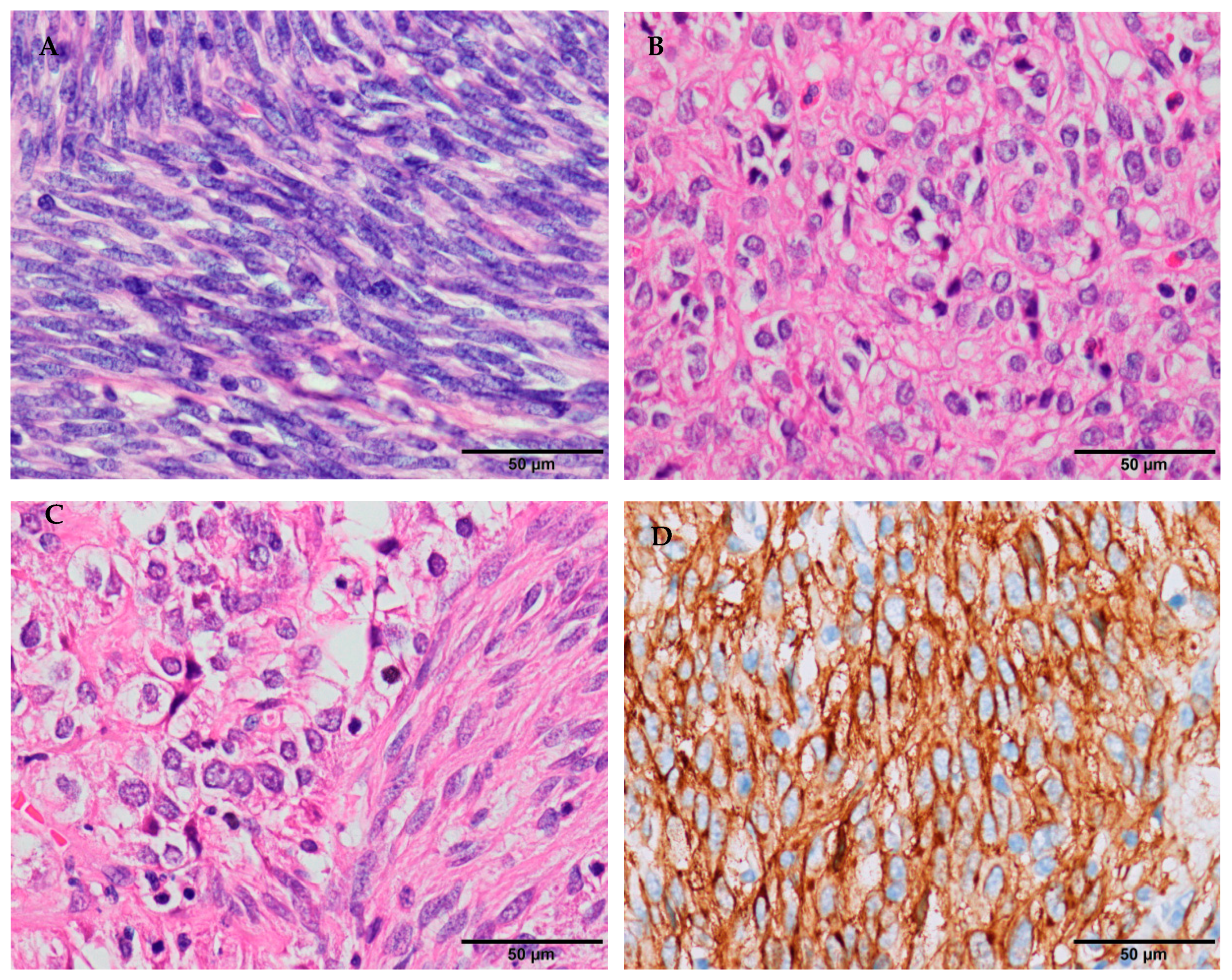
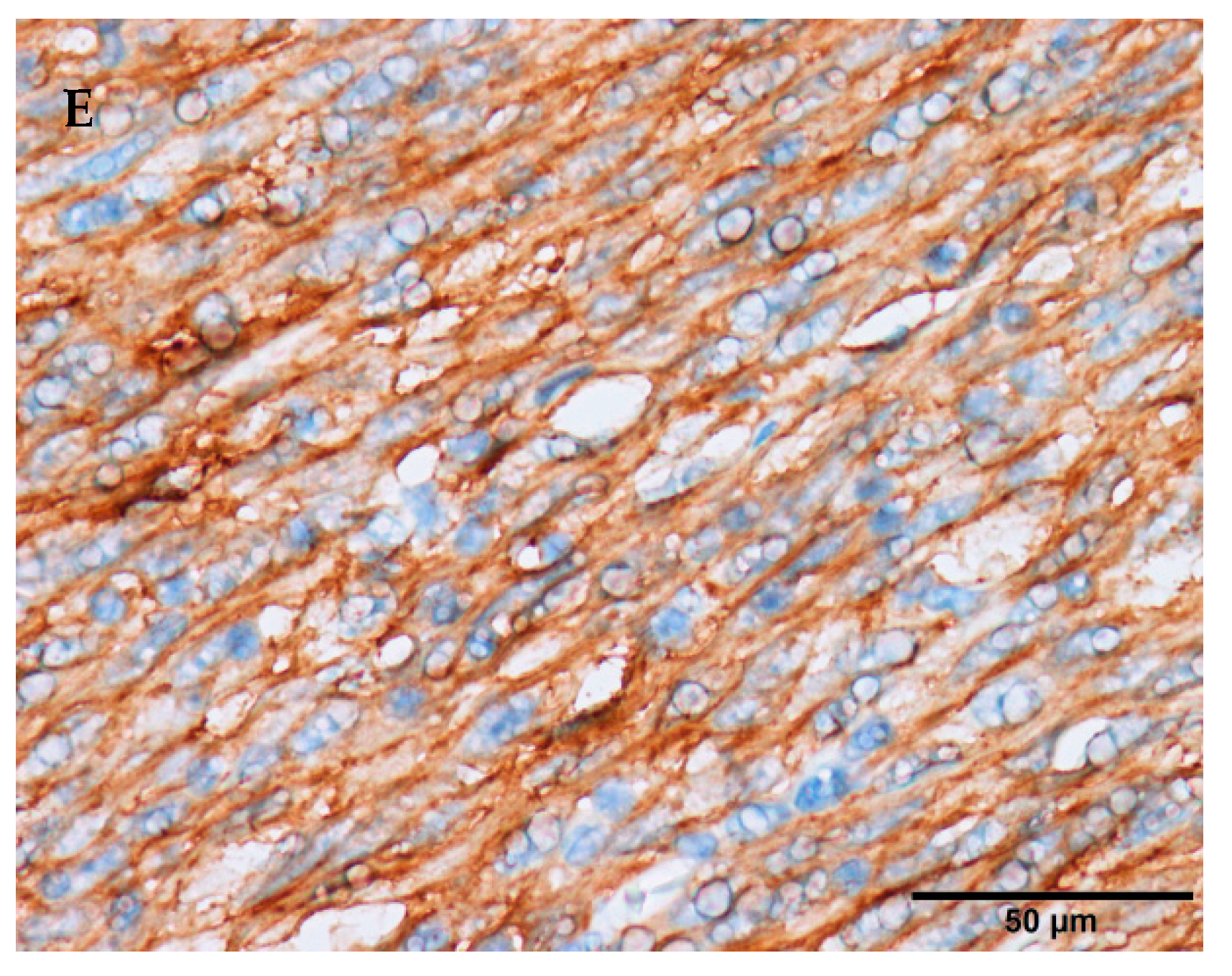
1.1. Clinical Features
1.2. Molecular Pathogenesis
1.3. Prognostic Factors and Risk Stratification
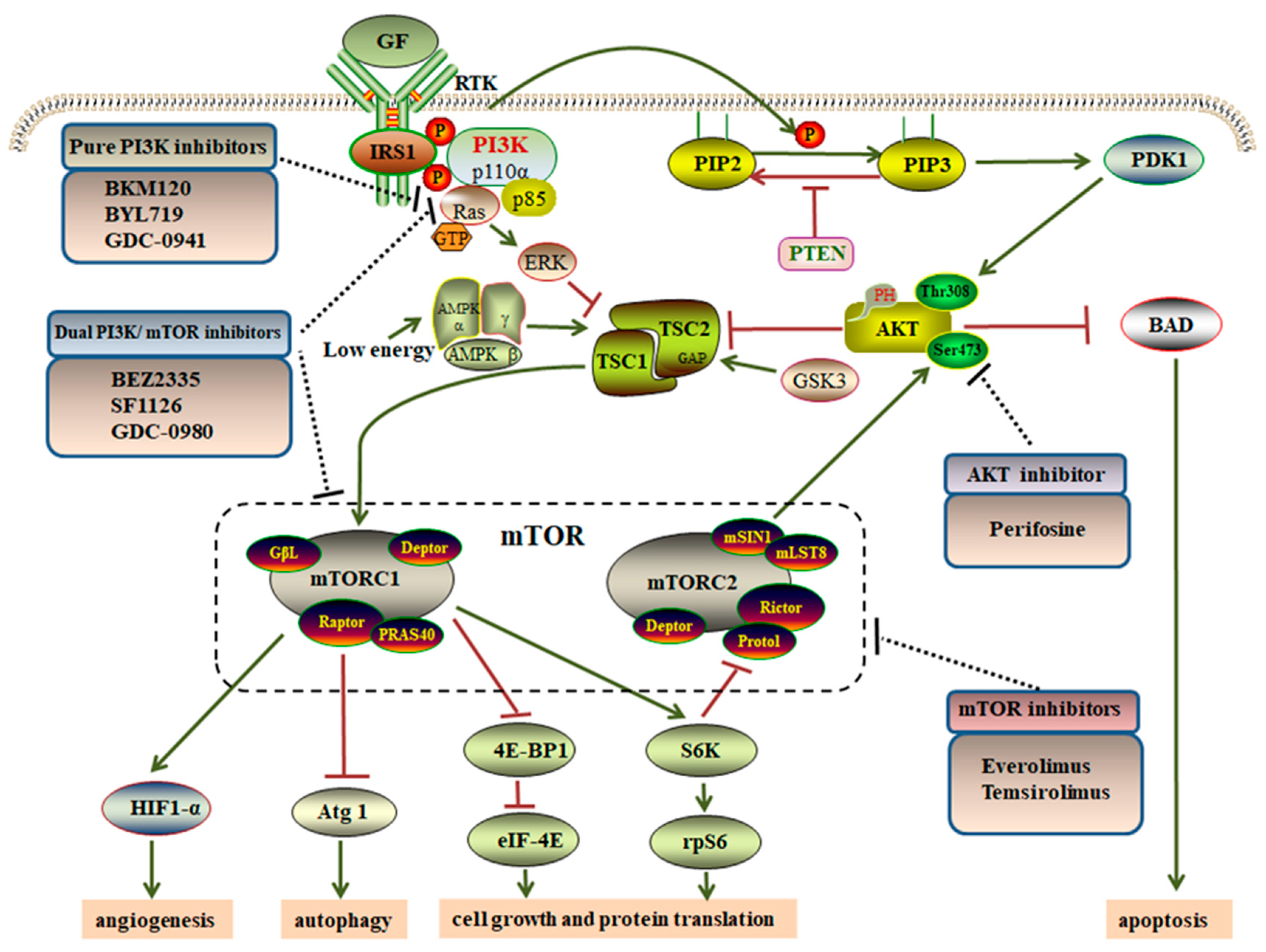
2. Overview of the PI3K/AKT/mTOR Pathway
3. Influence of the PI3K/AKT/mTOR Pathway on Proliferation, Apoptosis, Autophagy and Progression of GISTs
3.1. Influence on Cell Proliferation
3.2. Influence on Apoptosis
3.3. Influence on Autophagy
4. Targeting the PI3K/AKT/mTOR Pathway via GIST Therapeutics
4.1. Pure PI3K Inhibitors
4.2. AKT Inhibitors
4.3. mTOR Inhibitors
4.4. Dual PI3K/mTOR Inhibitors
5. eIFs and PI3K/AKT/mTOR Pathway
5.1. The Relationship between eIFs and the PI3K/AKT/mTOR Pathway
5.2. PI3K/AKT/mTOR-Regulated eIFs as a Potential Therapeutic Target in Tumors
6. Challenges and Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Agaimy, A.; Wünsch, P.H. Gastrointestinal stromal tumours: A regular origin in the muscularis propria, but an extremely diverse gross presentation. A review of 200 cases to critically re-evaluate the concept of so-called extra-gastrointestinal stromal tumours. Langenbecks Arch. Surg. 2006, 391, 322–329. [Google Scholar] [CrossRef]
- Søreide, K.; Sandvik, O.M.; Søreide, J.A.; Giljaca, V.; Jureckova, A.; Bulusu, V.R. Global epidemiology of gastrointestinal stromal tumors (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016, 40, 39–46. [Google Scholar] [CrossRef]
- Ud Din, N.; Ahmad, Z.; Arshad, H.; Idrees, R.; Kayani, N. Gastrointestinal Stromal Tumors: A Clinicopathologic and Risk Stratification Study of 255 Cases from Pakistan and Review of Literature. Asian Pac. J. Cancer Prev. 2015, 16, 4873–4880. [Google Scholar] [CrossRef] [PubMed]
- Gopie, P.; Mei, L.; Faber, A.C.; Grossman, S.R.; Smith, S.C.; Boikos, S.A. Classification of gastrointestinal stromal tumor syndromes. Endocr. Relat. Cancer 2018, 28, R49–R58. [Google Scholar] [CrossRef]
- Sorour, M.A.; Kassem, M.I.; Ghazal, A.-H.; El-Riwini, M.T.; Nasr, A.A. Gastrointestinal stromal tumors (GIST) related emergencies. Int. J. Surg. 2014, 12, 269–280. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Aubry, M.C.; Tazelaar, H.D.; Slezak, J.; Carney, J.A. Pulmonary chondroma: A tumor associated with Carney triad and different from pulmonary hamartoma. Am. J. Surg. Pathol. 2007, 31, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Boikos, S.A.; Xekouki, P.; Fumagalli, E.; Faucz, F.R.; Raygada, M.; Szarek, E.; Ball, E.; Kim, S.Y.; Miettinen, M.; Helman, L.J. Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur. J. Hum. Genet. 2016, 24, 569–573. [Google Scholar] [CrossRef]
- Kramer, K.; Siech, M.; Sträter, J.; Aschoff, A.J.; Henne-Bruns, D. GI hemorrhage with fulminant shock induced by jejunal gastrointestinal stromal tumor (GIST) coincident with duodenal neuroendocrine carcinoma (NET) + neurofibromatosis (NF)—Case report and review of the literature. Z. Gastroenterol. 2005, 43, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulou-Strauss, A.; Ronellenfitsch, U.; Cheng, C.; Pan, L.; Sachpekidis, C.; Hohenberger, P.; Henzler, T. Imaging therapy response of gastrointestinal stromal tumors (GIST) with FDG PET, CT and MRI: A systematic review. Clin. Transl. Imaging 2017, 5, 183–197. [Google Scholar] [CrossRef]
- Burkill, G.J.; Badran, M.; Al-Muderis, O.; MeirionThomas, J.; Judson, I.R.; Fisher, C.; Moskovic, E.C. Malignant Gastrointestinal Stromal Tumor: Distribution, Imaging Features, and Pattern of Metastatic Spread1. Radiology 2003, 226, 527–532. [Google Scholar] [CrossRef]
- Yin, Y.Q.; Liu, C.J.; Zhang, B.; Wen, Y.; Yin, Y. Association between CT imaging features and KIT mutations in small intestinal gastrointestinal stromal tumors. Sci. Rep. 2019, 9, 7257. [Google Scholar] [CrossRef]
- Shinagare, A.B.; Ip, I.K.; Lacson, R.; Ramaiya, N.H.; George, S.; Khorasani, R. Gastrointestinal stromal Tumor: Optimizing the Use of Cross-sectional Chest Imaging during Follow-up. Radiology 2015, 274, 395–404. [Google Scholar] [CrossRef]
- Kalkmann, J.; Zeile, M.; Antoch, G.; Berger, F.; Diederich, S.; Dinter, D.; Fink, C.; Janka, R.; Stattaus, J. Consensus report on the radiological management of patients with gastrointestinal stromal tumours (GIST): Recommendations of the German GIST Imaging Working Group. Cancer Imaging 2012, 12, 126–135. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, S.W. Performance of F-18 FDG PET/CT for predicting malignant potential of gastrointestinal stromal tumors: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2018, 33, 576–582. [Google Scholar] [CrossRef]
- Miettinen, M.; Sobin, L.H.; Lasota, J. Gastrointestinal Stromal Tumors of the Stomach: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 1765 Cases with Long-term Follow-up. Am. J. Surg. Pathol. 2005, 29, 52–68. [Google Scholar] [CrossRef]
- Apostolou, K.G.; Schizas, D.; Vavouraki, E.; Michalinos, A.; Tsilimigras, D.I.; Garmpis, N.; Damaskos, C.; Papalampros, A.; Liakakos, T. Clinicopathological and Molecular Factors, Risk Factors, Treatment Outcomes and Risk of Recurrence in Mesenteric and Retroperitoneal Extragastrointestinal Stromal Tumors. Anticancer Res. 2018, 38, 1903–1909. [Google Scholar] [CrossRef]
- Kim, K.H.; Nelson, S.D.; Kim, D.H.; Choi, K.U.; Kim, S.J.; Min, K.W.; Jang, K.S.; Paik, S.S.; Oh, Y.H.; Chae, S.W. Diagnostic relevance of overexpressions of PKC-θ and DOG-1 and KIT/PDGFRA gene mutations in extragastrointestinal stromal tumors: A Korean six-centers study of 28 cases. Anticancer Res. 2012, 32, 923–937. [Google Scholar]
- Hornick, J.L.; Fletcher, C.D. The Significance of KIT (CD117) in Gastrointestinal Stromal Tumors. Int. J. Surg. Pathol. 2004, 12, 93–97. [Google Scholar] [CrossRef]
- Novelli, M.; Rossi, S.; Rodriguez-Justo, M.; Taniere, P.; Seddon, B.; Toffolatti, L.; Sartor, C.; Hogendoorn, P.C.; Sciot, R.; Van Glabbeke, M. DOG1 and CD117 are the antibodies of choice in the diagnosis of gastrointestinal stromal tumours. Histopathology 2010, 57, 259–270. [Google Scholar] [CrossRef]
- Gomez-Pinilla, P.J.; Gibbons, S.J.; Bardsley, M.R.; Lorincz, A.; Pozo, M.J.; Pasricha, P.J.; Van de Rijn, M.; West, R.B.; Sarr, M.G.; Kendrick, M.L. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1370–G1381. [Google Scholar] [CrossRef]
- Miettinen, M.; Wang, Z.F.; Lasota, J. DOG1 Antibody in the Differential Diagnosis of Gastrointestinal Stromal Tumors: A Study of 1840 Cases. Am. J. Surg. Pathol. 2009, 33, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Von Mehren, M.; Joensuu, H. Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2018, 36, 136–143. [Google Scholar] [CrossRef]
- Rubin, B.P.; Singer, S.; Tsao, C.; Duensing, A.; Lux, M.L.; Ruiz, R.; Hibbard, M.K.; Chen, C.J.; Xiao, S.; Tuveson, D.A. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001, 61, 8118–8121. [Google Scholar]
- Joensuu, H.; Rutkowski, P.; Nishida, T.; Steigen, S.E.; Brabec, P.; Plank, L.; Nilsson, B.; Braconi, C.; Bordoni, A.; Magnusson, M.K. KIT and PDGFRA Mutations and the Risk of GI Stromal Tumor Recurrence. J. Clin. Oncol. 2015, 33, 634–642. [Google Scholar] [CrossRef]
- Yamamoto, H.; Oda, Y.; Kawaguchi, K.; Nakamura, N.; Takahira, T.; Tamiya, S.; Saito, T.; Oshiro, Y.; Ohta, M.; Yao, T. c-kit and PDGFRA mutations in extragastrointestinal stromal tumor (gastrointestinal stromal tumor of the soft tissue). Am. J. Surg. Pathol. 2004, 28, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Tabone-Eglinger, S.; Subra, F.; El Sayadi, H.; Alberti, L.; Tabone, E.; Michot, J.P.; Théou-Anton, N.; Lemoine, A.; Blay, J.Y.; Emile, J.F. KIT mutations induce intracellular retention and activation of an immature form of the KIT protein in gastrointestinal stromal tumors. Clin Cancer Res. 2008, 15, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; Van der Graaf, W.T.; Jones, R.L. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017, 13, 93–107. [Google Scholar] [CrossRef]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Cassier, P.A.; Fumagalli, E.; Rutkowski, P.; Schöffski, P.; Van Glabbeke, M.; Debiec-Rychter, M.; Emile, J.F.; Duffaud, F.; Martin-Broto, J.; Landi, B. Outcome of Patients with Platelet-Derived Growth Factor Receptor Alpha-Mutated Gastrointestinal Stromal Tumors in the Tyrosine Kinase Inhibitor Era. Clin. Cancer Res. 2012, 18, 4458–4464. [Google Scholar] [CrossRef] [PubMed]
- Settas, N.; Faucz, F.R.; Stratakis, C.A. Succinate dehydrogenase (SDH) deficiency, Carney triad and the epigenome. Mol. Cell. Endocrinol. 2018, 469, 107–111. [Google Scholar] [CrossRef]
- Li, J.; Ye, Y.; Wang, J.; Zhang, B.; Qin, S.; Shi, Y.; He, Y.; Liang, X.; Liu, X.; Zhou, Y. Chinese consensus guidelines for diagnosis and management of gastrointestinal stromal tumor. Chin. J. Cancer Res. 2017, 29, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Khoo, C.Y.; Chai, X.; Quek, R.; Teo, M.C.C.; Goh, B.K.P. Systematic review of current prognostication systems for primary gastrointestinal stromal tumors. Eur. J. Surg. Oncol. 2018, 44, 388–394. [Google Scholar] [CrossRef]
- Li, J.; Gong, J.F.; Wu, A.W.; Shen, L. Post-operative imatinib in patients with intermediate or high risk gastrointestinal stromal tumor. Eur. J. Surg. Oncol. 2011, 37, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Cavnar, M.J.; Seier, K.; Curtin, C.; Balachandran, V.P.; Coit, D.G.; Yoon, S.S.; Crago, A.M.; Strong, V.E.; Tap, W.D.; Gönen, M. Outcome of 1000 Patients With Gastrointestinal Stromal Tumor (GIST) Treated by Surgery in the Pre and Post-imatinib Eras. Ann. Surg. 2019. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.L.; Wang, J.C.; Zou, W.B.; Yao, D.K. Clinicopathological characteristics and prognostic factors of gastrointestinal stromal tumors in Chinese patients. Oncol. Lett. 2018, 16, 4905–4914. [Google Scholar] [CrossRef]
- DeMatteo, R.P.; Lewis, J.J.; Leung, D.; Mudan, S.S.; Woodruff, J.M.; Brennan, M.F. Two hundred gastrointestinal stromal tumors: Recurrence patterns and prognostic factors for survival. Ann. Surg. 2000, 231, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.; Roberts, P.J.; Heinz, D.; et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef]
- Van Glabbeke, M.; Verweij, J.; Casali, P.G.; Le Cesne, A.; Hohenberger, P.; Ray-Coquard, I.; Schlemmer, M.; van Oosterom, A.T.; Goldstein, D.; Sciot, R.; et al. Initial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: A European Organisation for Research and Treatment of Cancer-Italian Sarcoma Group-Australasian Gastrointestinal Trials Group study. J. Clin. Oncol. 2005, 23, 5795–5804. [Google Scholar] [CrossRef]
- Zhi, X.; Zhou, X.; Wang, W.; Xu, Z. Practical role of mutation analysis for imatinib treatment in patients with advanced gastrointestinal stromal tumors: A meta-analysis. PLoS ONE 2013, 8, e79275. [Google Scholar] [CrossRef]
- Wardelmann, E.; Losen, I.; Hans, V.; Neidt, I.; Speidel, N.; Bierhoff, E.; Heinicke, T.; Pietsch, T.; Büttner, R.; Merkelbach-Bruse, S. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int. J. Cancer 2003, 106, 887–895. [Google Scholar] [CrossRef]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Polish Clinical GIST Registry. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann. Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef]
- Qi, C.; Pan, F.; Li, J.; Li, Y.; Gao, J.; Shen, L. Analysis of biological characteristics and prognosis on gastrointestinal stromal tumor with PDGFRA gene mutation. Zhonghua Wei Chang Wai Ke Za Zhi 2018, 21, 1280–1284. (In Chinese) [Google Scholar] [PubMed]
- Lasota, J.; Dansonka-Mieszkowska, A.; Sobin, L.H.; Miettinen, M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab. Investig. 2004, 84, 874–883. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef] [PubMed]
- Ghoneum, A.; Said, N. PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers 2019, 11, 949. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Enya Chen, Y.C.; Burgess, M.; Mapp, S.; Mollee, P.; Gill, D.; Blumenthal, A.; Saunders, N.A. PI3K-p110δ contributes to antibody responses by macrophages in chronic lymphocytic leukemia. Leukemia 2020, 34, 451–461. [Google Scholar] [CrossRef]
- Rathinaswamy, M.K.; Burke, J.E. Class I phosphoinositide 3-kinase (PI3K) regulatory subunits and their roles in signaling and disease. Adv. Biol. Regul. 2020, 75, 100657. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Markman, B.; Atzori, F.; Pérez-García, J.; Tabernero, J.; Baselga, J. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann. Oncol. 2010, 21, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Klippel, A.; Kavanaugh, W.M.; Pot, D.; Williams, L.T. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol. Cell. Biol. 1997, 17, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Komander, D.; van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.N.; Walker, A.L.; Liu, Y.Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef]
- Vahidnezhad, H.; Youssefian, L.; Uitto, J. Molecular Genetics of the PI3K-AKT-mTOR Pathway in Genodermatoses: Diagnostic Implications and Treatment Opportunities. J. Investig. Dermatol. 2016, 136, 15–23. [Google Scholar] [CrossRef]
- Ma, L.; Teruya-Feldstein, J.; Bonner, P.; Bernardi, R.; Franz, D.N.; Witte, D.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 2007, 67, 7106–7112. [Google Scholar] [CrossRef]
- Ballif, B.A.; Roux, P.P.; Gerber, S.A.; MacKeigan, J.P.; Blenis, J.; Gygi, S.P. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc. Natl. Acad. Sci. USA 2005, 102, 667–672. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Lee, J.; Hong, E.M.; Kim, J.H.; Jung, J.H.; Park, S.W.; Koh, D.H.; Choi, M.H.; Jang, H.J.; Kae, S.H. Metformin Induces Apoptosis and Inhibits Proliferation through the AMP-Activated Protein Kinase and Insulin-like Growth Factor 1 Receptor Pathways in the Bile Duct Cancer Cells. J. Cancer 2019, 10, 1734–1744. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Wendel, H.G.; De Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Ata, R.; Antonescu, C.N. Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. Int. J. Mol. Sci. 2017, 18, 189. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef]
- Scott, R.C.; Juhász, G.; Neufeld, T.P. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 2007, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, S.; Lee, J.; Park, J.; Lee, G.; Kim, Y.; Kim, J.M.; Chung, J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007, 8, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Petrulea, M.S.; Plantinga, T.S.; Smit, J.W.; Georgescu, C.E.; Netea-Maier, R.T. PI3K/Akt/mTOR: A promising therapeutic target for non-medullary thyroid carcinoma. Cancer Treat. Rev. 2015, 41, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Duensing, A.; Demetri, G.D.; Fletcher, J.A. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene 2007, 26, 7560–7568. [Google Scholar] [CrossRef]
- Li, J.; Dang, Y.; Gao, J.; Li, Y.; Zou, J.; Shen, L. PI3K/AKT/mTOR pathway is activated after imatinib secondary resistance in gastrointestinal stromal tumors (GISTs). Med. Oncol. 2015, 32, 111. [Google Scholar] [CrossRef]
- Teng, Q.X.; Ashar, Y.V.; Gupta, P.; Gadee, E.; Fan, Y.F.; Reznik, S.E.; Wurpel, J.N.D.; Chen, Z.S. Revisiting mTOR inhibitors as anticancer agents. Drug Discov. Today 2019, 24, 2086–2095. [Google Scholar] [CrossRef]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef]
- Sápi, Z.; Füle, T.; Hajdu, M.; Matolcsy, A.; Moskovszky, L.; Márk, A.; Sebestyén, A.; Bodoky, G. The activated targets of mTOR signaling pathway are characteristic for PDGFRA mutant and wild-type rather than KIT mutant GISTs. Diagn. Mol. Pathol. 2011, 20, 22–33. [Google Scholar] [CrossRef]
- Pang, Y.; Xie, F.; Cao, H.; Wang, C.; Zhu, M.; Liu, X.; Lu, X.; Huang, T.; Shen, Y.; Li, K.; et al. Mutational inactivation of mTORC1 repressor gene DEPDC5 in human gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2019, 116, 22746–22753. [Google Scholar] [CrossRef]
- Li, C.F.; Fang, F.M.; Chen, Y.Y.; Liu, T.T.; Chan, T.C.; Yu, S.C.; Chen, L.T.; Huang, H.Y. Overexpressed Fatty Acid Synthase in Gastrointestinal Stromal Tumors: Targeting a Progression-Associated Metabolic Driver Enhances the Antitumor Effect of Imatinib. Clin. Cancer Res. 2017, 23, 4908–4918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kou, Y.W. Study of the expressions of p53 and bcl-2 genes, the telomerase activity and apoptosis in GIST patients. World J. Gastroenterol. 2007, 13, 2626–2628. [Google Scholar] [CrossRef]
- Ríos-Moreno, M.J.; Jaramillo, S.; Díaz-Delgado, M.; Sánchez-León, M.; Trigo-Sánchez, I.; Padillo, J.P.; Amérigo, J.; González-Cámpora, R. Differential activation of MAPK and PI3K/AKT/mTOR pathways and IGF1R expression in gastrointestinal stromal tumors. Anticancer Res. 2011, 31, 3019–3025. [Google Scholar]
- Ma, Y.Y.; Yu, S.; He, X.J.; Xu, Y.; Wu, F.; Xia, Y.J.; Guo, K.; Wang, H.J.; Ye, Z.Y.; Zhang, W.; et al. Involvement of c-KIT mutation in the development of gastrointestinal stromal tumors through proliferation promotion and apoptosis inhibition. OncoTargets Ther. 2014, 7, 637–643. [Google Scholar] [CrossRef]
- Ihle, M.A.; Trautmann, M.; Kuenstlinger, H.; Huss, S.; Heydt, C.; Fassunke, J.; Wardelmann, E.; Bauer, S.; Schildhaus, H.U.; Buettner, R.; et al. miRNA-221 and miRNA-222 induce apoptosis via the KIT/AKT signalling pathway in gastrointestinal stromal tumours. Mol. Oncol. 2015, 9, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Biray Avci, C.; Sezgin, B.; Goker Bagca, B.; Karci, H.B.; Gode, S. PI3K/AKT/mTOR pathway and autophagy regulator genes in paranasal squamous cell carcinoma metastasis. Mol. Biol. Rep. 2020, 47, 3641–3651. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Han, T.; Guo, M.; Gan, M.; Yu, B.; Tian, X.; Wang, J.B. TRIM59 regulates autophagy through modulating both the transcription and the ubiquitination of BECN1. Autophagy 2018, 14, 2035–2048. [Google Scholar] [CrossRef]
- Miselli, F.; Negri, T.; Gronchi, A.; Losa, M.; Conca, E.; Brich, S.; Fumagalli, E.; Fiore, M.; Casali, P.G.; Pierotti, M.A.; et al. Is autophagy rather than apoptosis the regression driver in imatinib-treated gastrointestinal stromal tumors? Transl. Oncol. 2008, 1, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Z.; Liu, H.; Jiang, S.; Wang, G.; Sun, L.; Li, J.; Wang, X.; Yu, S.; Huang, J.; et al. MicroRNA-30a targets BECLIN-1 to inactivate autophagy and sensitizes gastrointestinal stromal tumor cells to imatinib. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Gupta, A.; Roy, S.; Lazar, A.J.; Wang, W.L.; McAuliffe, J.C.; Reynoso, D.; McMahon, J.; Taguchi, T.; Floris, G.; Debiec-Rychter, M.; et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc. Natl. Acad. Sci. USA 2010, 107, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.S.; Yen, C.C.; Shih, N.Y.; Chiang, N.J.; Li, C.F.; Chen, L.T. Autophagy is involved in endogenous and NVP-AUY922-induced KIT degradation in gastrointestinal stromal tumors. Autophagy 2013, 9, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.S.; Chang, H.H.; Chiang, N.J.; Yen, C.C.; Li, C.F.; Chen, L.T. MTOR inhibition enhances NVP-AUY922-induced autophagy-mediated KIT degradation and cytotoxicity in imatinib-resistant gastrointestinal stromal tumors. Oncotarget 2014, 5, 11723–11736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, T.; Wang, W.; Piao, D. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumor after failure with imatinib and sunitinib treatment: A meta-analysis. Medicine 2017, 96, e8698. [Google Scholar] [CrossRef]
- Zhao, W.; Qiu, Y.; Kong, D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef]
- Maira, S.M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef]
- Van Looy, T.; Wozniak, A.; Floris, G.; Sciot, R.; Li, H.; Wellens, J.; Vanleeuw, U.; Fletcher, J.A.; Manley, P.W.; Debiec-Rychter, M.; et al. Phosphoinositide 3-kinase inhibitors combined with imatinib in patient-derived xenograft models of gastrointestinal stromal tumors: Rationale and efficacy. Clin. Cancer Res. 2014, 20, 6071–6082. [Google Scholar] [CrossRef]
- Floris, G.; Wozniak, A.; Sciot, R.; Li, H.; Friedman, L.; Van Looy, T.; Wellens, J.; Vermaelen, P.; Deroose, C.M.; Fletcher, J.A.; et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: Long-lasting responses after treatment withdrawal. Clin. Cancer Res. 2013, 19, 620–630. [Google Scholar] [CrossRef]
- Markman, B.; Dienstmann, R.; Tabernero, J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget 2010, 1, 530–543. [Google Scholar] [CrossRef]
- Shome, D.; Trent, J.; Espandar, L.; Hatef, E.; Araujo, D.M.; Song, C.D.; Kim, S.K.; Esmaeli, B. Ulcerative keratitis in gastrointestinal stromal tumor patients treated with perifosine. Ophthalmology 2008, 115, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S.; Yang, T.; Cibas, E.S.; Fletcher, J.A. An in vitro cytologic assay for evaluation of the KIT signaling pathway in gastrointestinal stromal tumors. Mod. Pathol. 2007, 20, 579–583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pantaleo, M.A.; Nicoletti, G.; Nanni, C.; Gnocchi, C.; Landuzzi, L.; Quarta, C.; Boschi, S.; Nannini, M.; Di Battista, M.; Castellucci, P.; et al. Preclinical evaluation of KIT/PDGFRA and mTOR inhibitors in gastrointestinal stromal tumors using small animal FDG PET. J. Exp. Clin. Cancer Res. 2010, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Reichardt, P.; Blay, J.Y.; Dumez, H.; Morgan, J.A.; Ray-Coquard, I.; Hollaender, N.; Jappe, A.; Demetri, G.D. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann. Oncol. 2010, 21, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Santos, C.J.; Uzel, G.; Rosenzweig, S.D. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J. Allergy Clin. Immunol. 2019, 143, 1676–1687. [Google Scholar] [CrossRef]
- Quattrone, A.; Wozniak, A.; Dewaele, B.; Floris, G.; Vanspauwen, V.; Van Looy, T.; Schöffski, P.; Rutkowski, P.; Sciot, R.; Debiec-Rychter, M. Frequent mono-allelic loss associated with deficient PTEN expression in imatinib-resistant gastrointestinal stromal tumors. Mod. Pathol. 2014, 27, 1510–1520. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Chiorean, E.G.; Harris, W.; Von Hoff, D.D.; Younger, A.; RensvoldF Cordova, D.M.; Qi, W.; Shelton, C.F.; Becker, M.D.; Garlich, J.R.; et al. Phase I study of the multikinase prodrug SF1126 in solid tumors and B-cell malignancies. J. Clin. Oncol. 2011, 29 (Suppl. 15), 3015, Abstract. [Google Scholar] [CrossRef]
- Wagner, A.J.; Bendell, J.C.; Dolly, S.; Morgan, J.A.; Ware, J.A.; FredricksonK, E.; Mazina, J.; Lauchle, J.O.; Burris, H.A.; De Bono, J.S. A frst-in-human, phase l study to evaluate the dual PI3K/mTOR inhibitor GDC-0980 administered QD in patients with advanced solid tumors or non-Hodgkin’s lymphoma. J. Clin. Oncol. 2011, 29 (Suppl. 15), 3020, Abstract. [Google Scholar] [CrossRef]
- Hershey, J.W.; Sonenberg, N.; Mathews, M.B. Principles of translational control: An overview. Cold Spring Harb. Perspect. Biol. 2012, 4, a011528. [Google Scholar] [CrossRef]
- Delaidelli, A.; Jan, A.; Herms, J.; Sorensen, P.H. Translational control in brain pathologies: Biological significance and therapeutic opportunities. Acta Neuropathol. 2019, 137, 535–555. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.K.; Bressler, K.; Patel, H.; Balasingam, N.; Thakor, N. Role of Eukaryotic Initiation Factors during Cellular Stress and Cancer Progression. J. Nucleic Acids 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Seeboeck, R.; Sarne, V.; Haybaeck, J. Current Coverage of the mTOR Pathway by Next-Generation Sequencing Oncology Panels. Int. J. Mol. Sci. 2019, 20, 690. [Google Scholar] [CrossRef] [PubMed]
- Masvidal, L.; Hulea, L.; Furic, L.; Topisirovic, I.; Larsson, O. mTOR-sensitive translation: Cleared fog reveals more trees. RNA Biol. 2017, 14, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Naineni, S.K.; Itoua Maïga, R.; Cencic, R.; Putnam, A.A.; Amador, L.A.; Rodriguez, A.D.; Jankowsky, E.; Pelletier, J. A comparative study of small molecules targeting eIF4A. RNA 2020, 26, 541–549. [Google Scholar] [CrossRef]
- Chen, K.; Yang, J.; Li, J.; Wang, X.; Chen, Y.; Huang, S.; Chen, J.L. eIF4B is a convergent target and critical effector of oncogenic Pim and PI3K/Akt/mTOR signaling pathways in Abl transformants. Oncotarget 2016, 7, 10073–10089. [Google Scholar] [CrossRef]
- Cencic, R.; Robert, F.; Galicia-Vázquez, G.; Malina, A.; Ravindar, K.; Somaiah, R.; Pierre, P.; Tanaka, J.; Deslongchamps, P.; Pelletier, J. Modifying chemotherapy response by targeted inhibition of eukaryotic initiation factor 4A. Blood Cancer J. 2013, 3, e128. [Google Scholar] [CrossRef]
- Golob-Schwarzl, N.; Krassnig, S.; Toeglhofer, A.M.; Park, Y.N.; Gogg-Kamerer, M.; Vierlinger, K.; Schröder, F.; Rhee, H.; Schicho, R.; Fickert, P.; et al. New liver cancer biomarkers: PI3K/AKT/mTOR pathway members and eukaryotic translation initiation factors. Eur. J. Cancer 2017, 83, 56–70. [Google Scholar] [CrossRef]
- Tapia, O.; Riquelme, I.; Leal, P.; Sandoval, A.; Aedo, S.; Weber, H.; Letelier, P.; Bellolio, E.; Villaseca, M.; Garcia, P.; et al. The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch. 2014, 465, 25–33. [Google Scholar] [CrossRef]
- Wang, L.; Wen, X.; Luan, F.; Fu, T.; Gao, C.; Du, H.; Guo, T.; Han, J.; Huang, F.L.; Cheng, X.; et al. EIF3B is associated with poor outcomes in gastric cancer patients and promotes cancer progression via the PI3K/AKT/mTOR signaling pathway. Cancer Manag. Res. 2019, 11, 7877–7891. [Google Scholar] [CrossRef] [PubMed]
- Golob-Schwarzl, N.; Schweiger, C.; Koller, C.; Krassnig, S.; Gogg-Kamerer, M.; Gantenbein, N.; Toeglhofer, A.M.; Wodlej, C.; Bergler, H.; Pertschy, B.; et al. Separation of low and high grade colon and rectum carcinoma by eukaryotic translation initiation factors 1, 5 and 6. Oncotarget 2017, 8, 101224–101243. [Google Scholar] [CrossRef]
- Yan, L.; Zou, L.; Zhao, W.; Wang, Y.; Liu, B.; Yao, H.; Yu, H. Clinicopathological significance of c-KIT mutation in gastrointestinal stromal tumors: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 13718. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Zalcberg, J.; Le Cesne, A.; Reichardt, P.; Blay, J.Y.; Lindner, L.H.; Judson, I.R.; Schöffski, P.; Leyvraz, S.; Italiano, A.; et al. European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group. Ten-Year Progression-Free and Overall Survival in Patients with Unresectable or Metastatic GI Stromal Tumors: Long-Term Analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group Intergroup Phase III Randomized Trial on Imatinib at Two Dose Levels. J. Clin. Oncol. 2017, 35, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006, 130, 1466–1478. [Google Scholar] [CrossRef]
- Napolitano, A.; Vincenzi, B. Secondary KIT mutations: The GIST of drug resistance and sensitivity. Br. J. Cancer 2019, 120, 577–578. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Demetri, G.D.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D.; Van den Abbeele, A.D.; Corless, C.L.; Antonescu, C.R.; George, S.; Morgan, J.A.; Chen, M.H.; et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin. Cancer Res. 2009, 15, 5902–5909. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Serrano, C.; Mariño-Enríquez, A.; Tao, D.L.; Ketzer, J.; Eilers, G.; Zhu, M.; Yu, C.; Mannan, A.M.; Rubin, B.P.; Demetri, G.D.; et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2019, 120, 612–620. [Google Scholar] [CrossRef]
- Patel, S. Exploring novel therapeutic targets in GIST: Focus on the PI3K/Akt/mTOR pathway. Curr. Oncol. Rep. 2013, 15, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Kasireddy, V.; von Mehren, M. Emerging drugs for the treatment of gastrointestinal stromal tumour. Expert Opin. Emerg. Drugs 2017, 22, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Kalfusová, A.; Kodet, R. Molecular mechanisms of primary and secondary resistance, molecular-genetic features and characteristics of KIT/PDGFRA non-mutated GISTs. Ceskoslovenska Patol. 2017, 53, 167–173. [Google Scholar]
- Quan, P.; Moinfar, F.; Kufferath, I.; Absenger, M.; Kueznik, T.; Denk, H.; Zatloukal, K.; Haybaeck, J. Effects of targeting endometrial stromal sarcoma cells via histone deacetylase and PI3K/AKT/mTOR signaling. Anticancer Res. 2014, 34, 2883–2897. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Feature | Percentage |
|---|---|
| Pathological type | |
| Spindle cell type | 70% |
| Epithelioid cell type | 17% |
| Mixed, spindle-epithelioid type | 13% |
| Immunophenotype | |
| CD117-positivity | More than 90% |
| DOG1-positivity | 96% |
| Genotype | Percentage |
|---|---|
| KIT mutation | |
| KIT exon 11 mutation | 90% |
| KIT exon 9 mutation | 8% |
| KIT exon 13 mutation | 1% |
| KIT exon 17 mutation | 1% |
| PDGFRA | 5–8% |
| No KIT or PDGFRA mutation (including SDH-deficiency and non-SDH deficient) | 10–15% |
| Drug | Trial Phase | Target | Condition | Status | Trial Number |
|---|---|---|---|---|---|
| BYL719+ Imatinib | Phase Ib | c-KIT + PI3K p110α | Third-line GISTs | Recruiting | NCT01735968 |
| BKM120+Imatinib | Phase Ib | c-KIT + PI3K Class I | Third-line GISTs | Recruiting | NCT01468488 |
| RAD001+ Imatinib | Phase II | c-KIT + mTOR | Progressive GISTs | ongoing | NCT00510354 |
| RAD001+ Imatinib | Phase I/II | c-KIT + mTOR | Resistant GISTs | Completed | NCT01275222 |
| Temsirolimus | Phase II | mTOR | GISTs | Completed | NCT00087074 |
| Perifosine+ Imatinib | Phase II | c-KIT + AKT | Resistant GISTs | Completed | NCT00455559 |
| Perifosine+ Sunitinib | Phase I/II | c-KIT + AKT | Advanced GISTs | Completed | NCT00399052 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, Y.; Haybaeck, J.; Yang, Z. Therapeutic Potential of PI3K/AKT/mTOR Pathway in Gastrointestinal Stromal Tumors: Rationale and Progress. Cancers 2020, 12, 2972. https://doi.org/10.3390/cancers12102972
Duan Y, Haybaeck J, Yang Z. Therapeutic Potential of PI3K/AKT/mTOR Pathway in Gastrointestinal Stromal Tumors: Rationale and Progress. Cancers. 2020; 12(10):2972. https://doi.org/10.3390/cancers12102972
Chicago/Turabian StyleDuan, Yi, Johannes Haybaeck, and Zhihui Yang. 2020. "Therapeutic Potential of PI3K/AKT/mTOR Pathway in Gastrointestinal Stromal Tumors: Rationale and Progress" Cancers 12, no. 10: 2972. https://doi.org/10.3390/cancers12102972
APA StyleDuan, Y., Haybaeck, J., & Yang, Z. (2020). Therapeutic Potential of PI3K/AKT/mTOR Pathway in Gastrointestinal Stromal Tumors: Rationale and Progress. Cancers, 12(10), 2972. https://doi.org/10.3390/cancers12102972