Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning
2.2. Mice and Retroviral Infections of Primary Mouse Bone Marrow Cells
2.3. Proliferation Assay
2.4. Apoptosis Assay
2.5. Lipid Nanoparticle (LNPs)/siRNA Formulation
2.6. RNA Extraction, cDNA Synthesis, and Quantitative RT-PCR
2.7. Gene Expression Profiling and Analysis
2.8. Molecular Mutation Analysis
2.9. Statistical Analysis
3. Results
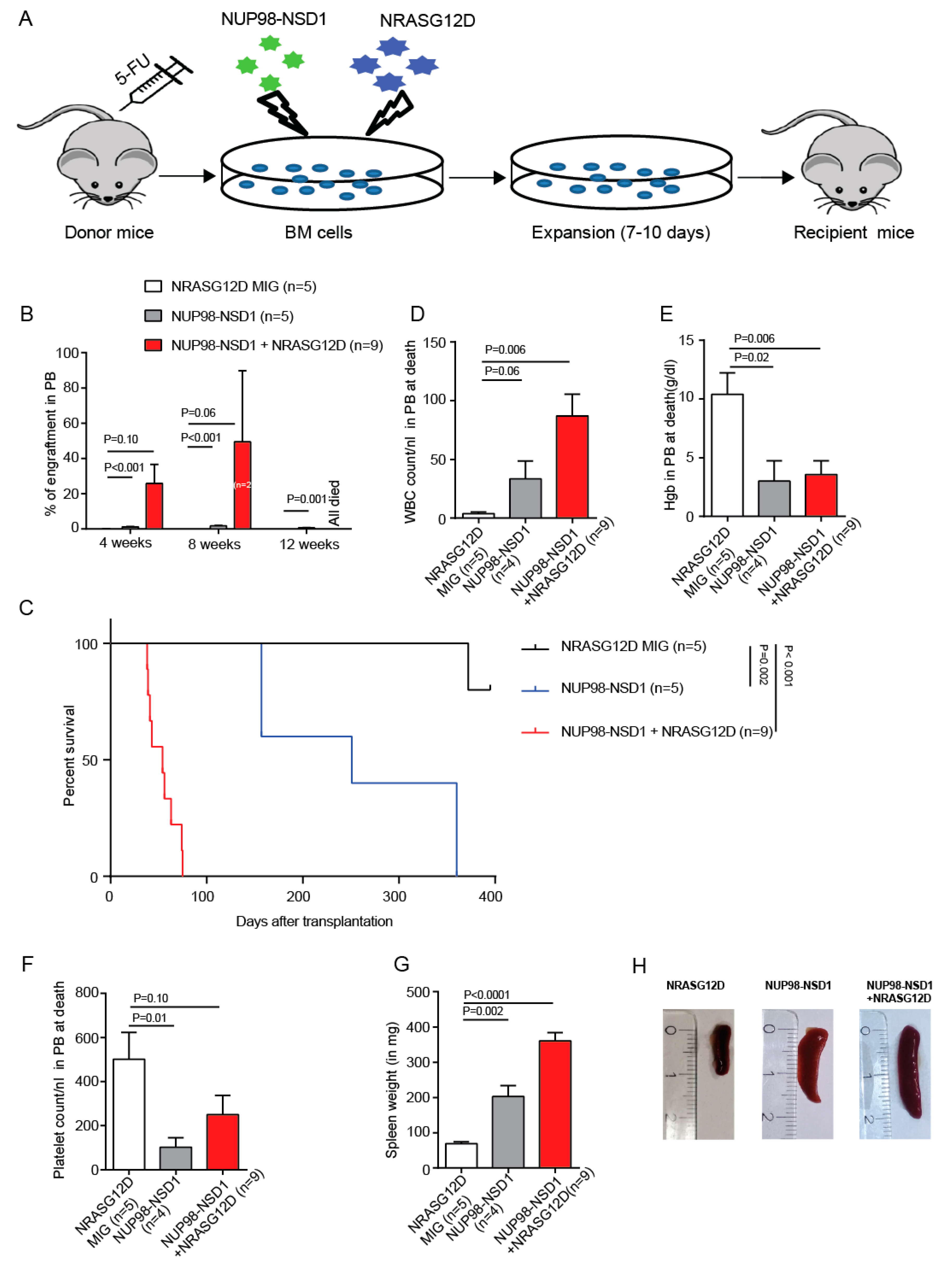
3.1. NUP98-NSD1 Immortalizes Murine Hematopoietic Cells and Cooperates with Mutated NRASG12D In Vitro
3.2. NUP98-NSD1 Induces a Long-Latency AML and Cooperates with NRASG12D to Induce an Aggressive AML In Vivo
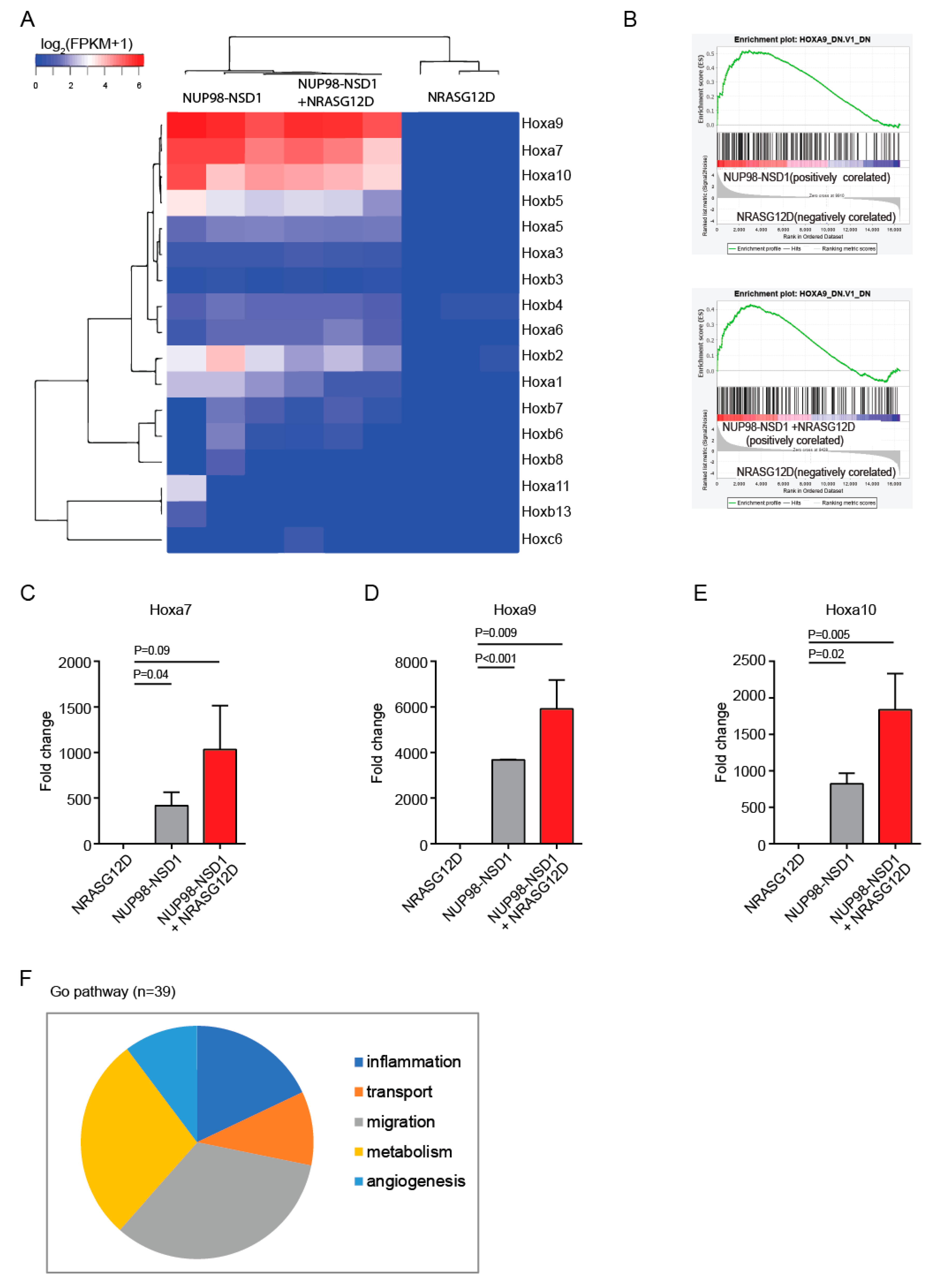
3.3. Common and Distinct Pathways Are Regulated by NUP98-NSD1 and NRASG12D
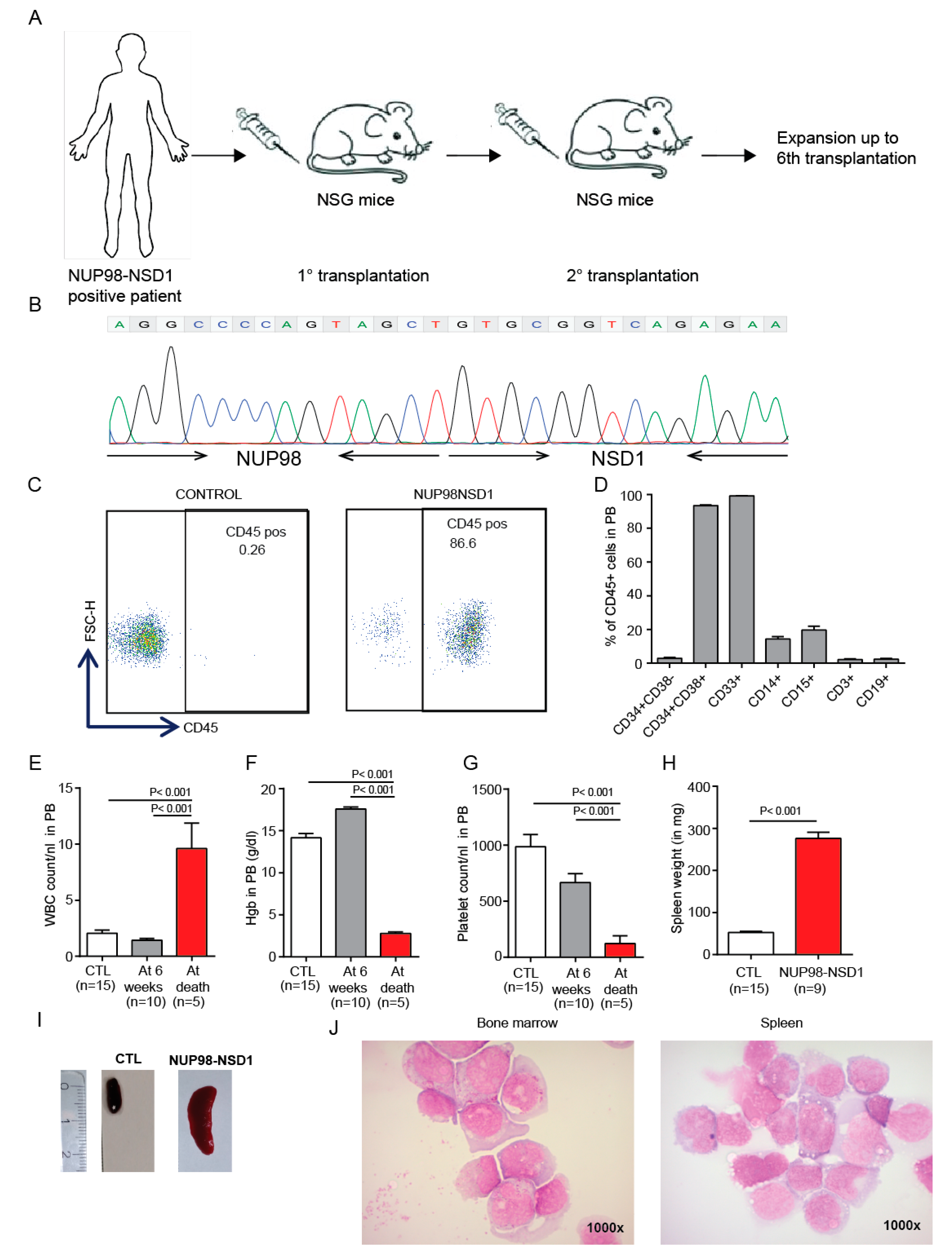
3.4. Establishment and Characterization of a NUP98-NSD1 Patient-Derived Xenograft (PDX) Model
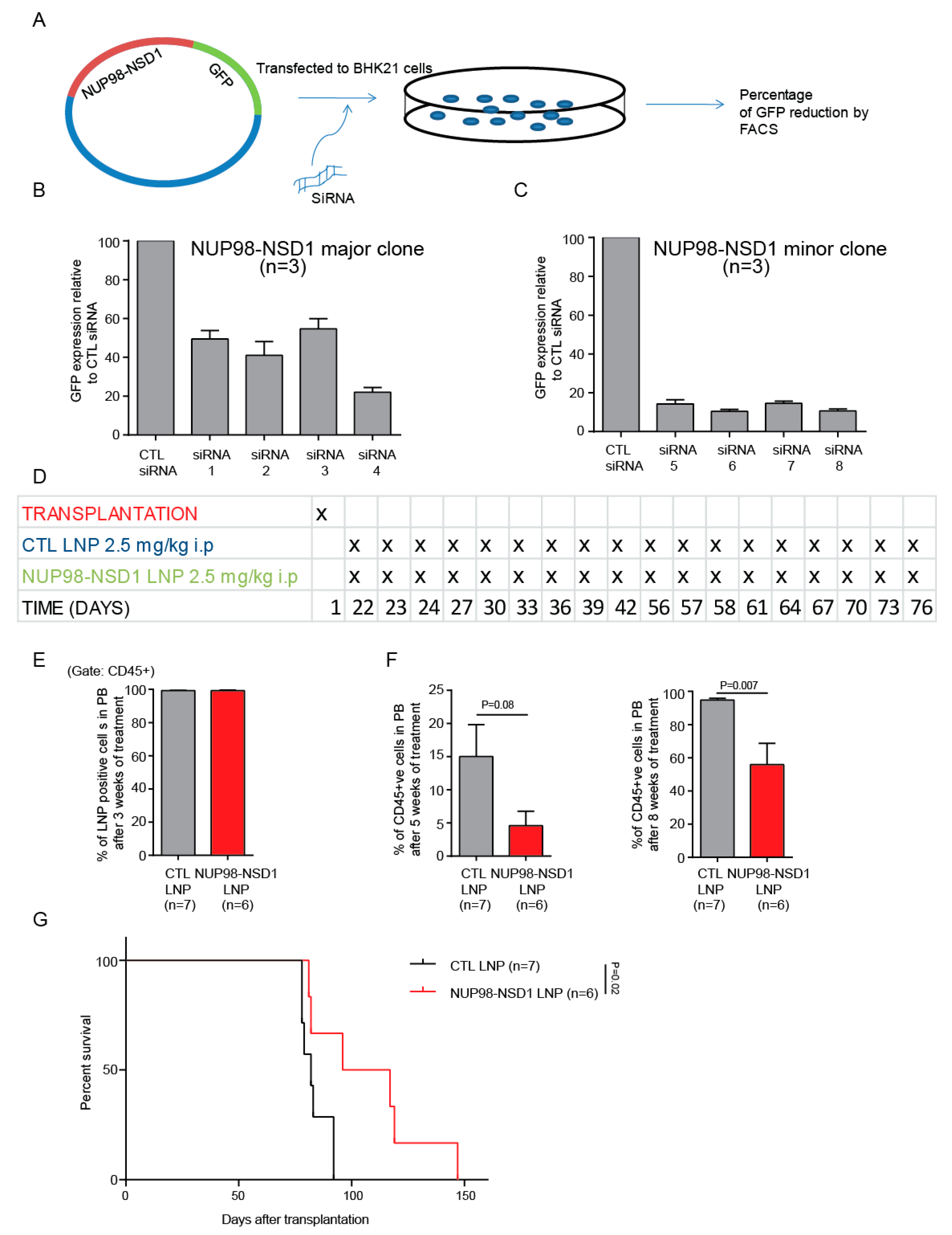
3.5. Treatment of the NUP98-NSD1 PDX Model with NUP98-NSD1 siRNA-LNPs Prolongs Survival of Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Heuser, M.; Ofran, Y.; Boissel, N.; Mauri, S.B.; Craddock, C.; Janssen, J.; Wierzbowska, A.; Buske, C. ESMO Guidelines Committee Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 697–712. [Google Scholar] [CrossRef]
- Arindrarto, W.; Borràs, D.M.; De Groen, R.A.L.; Berg, R.R.V.D.; Locher, I.J.; Van Diessen, S.A.M.E.; Van Der Holst, R.; Van Der Meijden, E.D.; Honders, M.W.; De Leeuw, R.H.; et al. Comprehensive diagnostics of acute myeloid leukemia by whole transcriptome RNA sequencing. Leukemia 2020. Advance Online Publication. [Google Scholar] [CrossRef]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef]
- Struski, S.; Lagarde, S.; Bories, P.; Puiseux, C.; Prade, N.; Cuccuini, W.; Pages, M.-P.; Bidet, A.; Gervais, C.; Lafage-Pochitaloff, M.; et al. NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 2016, 31, 565–572. [Google Scholar] [CrossRef]
- Hollink, I.H.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T.; Pratcorona, M.; Abbas, S.; Kuipers, J.E.; van Galen, J.F.; Beverloo, H.B.; Sonneveld, E.; Kaspers, G.J.J.; et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011, 118, 3645–3656. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kölking, B.; Hollink, I.H.I.; Damm, F.; Van Den Heuvel-eibrink, M.M.; Zwaan, C.M.; Bug, G.; Ottmann, O.; Wagner, K.; Morgan, M.; et al. Analysis of NUP98/NSD1 translocations in adult AML and MDS patients. Leukemia 2013, 27, 750–754. [Google Scholar] [CrossRef]
- Lavallée, V.-P.; Lemieux, S.; Boucher, G.; Gendron, P.; Boivin, I.; Girard, S.; Hébert, J.; Sauvageau, G. Identification of MYC mutations in acute myeloid leukemias with NUP98–NSD1 translocations. Leukemia 2016, 30, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Fasan, A.; Haferlach, C.; Alpermann, T.; Kern, W.; Haferlach, T.; Schnittger, S. A rare but specific subset of adult AML patients can be defined by the cytogenetically cryptic NUP98–NSD1 fusion gene. Leukemia 2012, 27, 245–2488. [Google Scholar] [CrossRef] [PubMed]
- Shiba, N.; Ichikawa, H.; Taki, T.; Park, M.-J.; Jo, A.; Mitani, S.; Kobayashi, T.; Shimada, A.; Sotomatsu, M.; Arakawa, H.; et al. NUP98-NSD1gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosom. Cancer 2013, 52, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Niktoreh, N.; Walter, C.; Zimmermann, M.; Von Neuhoff, C.; Von Neuhoff, N.; Rasche, M.; Waack, K.; Creutzig, U.; Hanenberg, H.; Reinhardt, D. Mutated WT1, FLT3-ITD, and NUP98-NSD1 Fusion in Various Combinations Define a Poor Prognostic Group in Pediatric Acute Myeloid Leukemia. J. Oncol. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ni, J.B.; Wang, X.Y.; Dai, Y.; Ma, X.L.; Su, Y.C.; Gao, Y.Y.; Chen, X.; Yuan, L.L.; Liu, H.X. Genetic characteristics and clinical outcomes of pediatric acute myeloid leukemia with NUP98-NSD1 fusion gene. Zhonghua Yi Xue Za Zhi 2019, 99, 2820–2825. [Google Scholar] [PubMed]
- Kivioja, J.L.; Martí, J.M.L.; Kumar, A.; Kontro, M.; Edgren, H.; Parsons, A.; Lundán, T.; Wolf, M.; Porkka, K.; Heckman, C.A. Chimeric NUP98–NSD1 transcripts from the cryptic t(5;11)(q35.2;p15.4) in adult de novo acute myeloid leukemia. Leuk. Lymphoma 2017, 59, 725–7322. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.M.; Hetzer, M.W. The role of Nup98 in transcription regulation in healthy and diseased cells. Trends Cell Biol. 2012, 23, 112–117. [Google Scholar] [CrossRef]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef]
- Bennett, R.L.; Swaroop, A.; Troche, C.; Licht, J.D. The Role of Nuclear Receptor–Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026708. [Google Scholar] [CrossRef]
- Wang, G.G.; Song, J.; Wang, Z.; Dormann, H.L.; Casadio, F.; Li, H.; Luo, J.-L.; Patel, D.J.; Allis, C.D. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009, 459, 847–851. [Google Scholar] [CrossRef]
- Yassin, E.R.; Abdul-Nabi, A.M.; Takeda, A.; Yaseen, N.R. Effects of the NUP98–DDX10 oncogene on primary human CD34+ cells: Role of a conserved helicase motif. Leukemia 2010, 24, 1001–1011. [Google Scholar] [CrossRef]
- Pineault, N.; Buske, C.; Feuring-Buske, M.; Abramovich, C.; Rosten, P.; Hogge, D.; Aplan, P.D.; Humphries, R.K. Induction of acute myeloid leukemia in mice by the human leukemia-specific fusion gene NUP98-HOXD13 in concert with Meis1. Blood 2003, 101, 4529–4538. [Google Scholar] [CrossRef]
- Wang, K.; Sanchez-Martin, M.; Wang, X.; Knapp, K.M.; Koche, R.; Vu, L.; Nahas, M.K.; He, J.; Hadler, M.; Stein, E.M.; et al. Patient-derived xenotransplants can recapitulate the genetic driver landscape of acute leukemias. Leukemia 2016, 31, 151–158. [Google Scholar] [CrossRef]
- Sharma, A.; Jyotsana, N.; Gabdoulline, R.; Heckl, D.; Kuchenbauer, F.; Slany, R.K.; Ganser, A.; Heuser, M. Meningioma 1 is indispensable for mixed lineage leukemia-rearranged acute myeloid leukemia. Haematologica 2019, 105, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Beutel, G.; Krauter, J.; Döhner, K.; Von Neuhoff, N.; Schlegelberger, B.; Ganser, A. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood 2006, 108, 3898–3905. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; González, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef]
- Heuser, M.; Gabdoulline, R.; Löffeld, P.; Dobbernack, V.; Kreimeyer, H.; Pankratz, M.; Flintrop, M.; Liebich, A.; Klesse, S.; Panagiota, V.; et al. Individual outcome prediction for myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia from MDS after allogeneic hematopoietic cell transplantation. Ann. Hematol. 2017, 96, 1361–1372. [Google Scholar] [CrossRef]
- Pineault, N.; Helgason, C.D.; Lawrence, H.J.; Humphries, R.K. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp. Hematol. 2002, 30, 49–57. [Google Scholar] [CrossRef]
- Keppler, D. Towards novel anti-cancer strategies based on cystatin function. Cancer Lett. 2006, 235, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Thanasopoulou, A.; Tzankov, A.; Schwaller, J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica 2014, 99, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Slape, C.I.; Liu, L.Y.; Beachy, S.; Aplan, P.D. Leukemic transformation in mice expressing a NUP98-HOXD13 transgene is accompanied by spontaneous mutations in Nras, Kras, and Cbl. Blood 2008, 112, 2017–2019. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, L.; Argiropoulos, B.; Pineault, N.; Abramovich, C.; Sly, L.M.; Krystal, G.; Wan, A.; Humphries, R.K. The Flt3 receptor tyrosine kinase collaborates with NUP98-HOX fusions in acute myeloid leukemia. Blood 2006, 108, 1030–1036. [Google Scholar] [CrossRef]
- Greenblatt, S.; Li, L.; Slape, C.; Nguyen, B.; Novak, R.; Duffield, A.; Huso, D.; Desiderio, S.; Borowitz, M.J.; Aplan, P.; et al. Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood 2012, 119, 2883–2894. [Google Scholar] [CrossRef]
- Drenberg, C.D.; Buelow, D.R.; Pounds, S.B.; Wang, Y.-D.; Finkelstein, D.; Rahija, R.J.; Shurtleff, S.A.; Rubnitz, J.E.; Inaba, H.; Gruber, T.A.; et al. Transcriptome profiling of patient derived xenograft models established from pediatric acute myeloid leukemia patients confirm maintenance of FLT3-ITD mutation. Leuk. Lymphoma 2016, 58, 247–250. [Google Scholar] [CrossRef]
- Grossmann, V.; Schnittger, S.; Poetzinger, F.; Kohlmann, A.; Stiel, A.; Eder, C.; Fasan, A.; Kern, W.; Haferlach, T.; Haferlach, C. High incidence of RAS signalling pathway mutations in MLL-rearranged acute myeloid leukemia. Leukemia 2013, 27, 1933–1936. [Google Scholar] [CrossRef]
- Krauth, M.T.; Eder, C.; Alpermann, T.; Bacher, U.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T.; Schnittger, S. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: Frequency and impact on clinical outcome. Leukemia 2014, 28, 1449–1458. [Google Scholar] [CrossRef]
- Paschka, P.; Du, J.; Schlenk, R.F.; Gaidzik, V.I.; Bullinger, L.; Corbacioglu, A.; Späth, D.; Kayser, S.; Schlegelberger, B.; Krauter, J.; et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): A study of the German-Austrian AML Study Group (AMLSG). Blood 2013, 121, 170–177. [Google Scholar] [CrossRef]
- Kuchenbauer, F.; Schnittger, S.; Look, T.; Gilliland, G.; Tenen, D.; Haferlach, T.; Hiddemann, W.; Buske, C.; Schoch, C. Identification of additional cytogenetic and molecular genetic abnormalities in acute myeloid leukaemia with t(8;21)/AML1-ETO. Br. J. Haematol. 2006, 134, 616–619. [Google Scholar] [CrossRef]
- Lin, Y.-W.; Slape, C.I.; Zhang, Z.; Aplan, P.D. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood 2005, 106, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Valerio, D.G.; Eisold, M.E.; Sinha, A.; Koche, R.P.; Hu, W.; Chen, C.-W.; Chu, S.H.; Brien, G.L.; Park, C.Y.; et al. NUP98 Fusion Proteins Interact with the NSL and MLL1 Complexes to Drive Leukemogenesis. Cancer Cell 2016, 30, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.M.; McCloskey, A.; Shokhirev, M.N.; Benner, C.; Rathore, A.; Hetzer, M.W.; Shokirev, M.N. Nup98 recruits the Wdr82–Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes Dev. 2017, 31, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.; Hess, J.L. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Kennedy, A.; Zhou, X.; Radtke, I.; Phillips, L.A.; Shurtleff, S.A.; Downing, J.R. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia 2007, 21, 2000–2009. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Caudell, D.; Zhang, Z.; Chung, Y.J.; Aplan, P.D. Expression of a CALM-AF10 fusion gene leads to Hoxa cluster overexpression and acute leukemia in transgenic mice. Cancer Res. 2007, 67, 8022–8031. [Google Scholar] [CrossRef]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Scherr, M.; Kuchenbauer, F.; Sajti, L.; Barchanski, A.; Lindner, R.; Noyan, F.; Sühs, K.-W.; et al. RNA interference efficiently targets human leukemia driven by a fusion oncogene in vivo. Leukemia 2017, 32, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Budida, R.; Scherr, M.; Kuchenbauer, F.; Lindner, R.; Noyan, F.; Sühs, K.-W.; Stangel, M.; et al. Lipid nanoparticle-mediated siRNA delivery for safe targeting of human CML in vivo. Ann. Hematol. 2019, 98, 1905–1918. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef]
- Cummins, K.D.; Gill, S. Chimeric antigen receptor T-cell therapy for acute myeloid leukemia: How close to reality? Haematologica 2019, 104, 1302–1308. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanty, S.; Jyotsana, N.; Sharma, A.; Kloos, A.; Gabdoulline, R.; Othman, B.; Lai, C.K.; Schottmann, R.; Mandhania, M.; Schmoellerl, J.; et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia. Cancers 2020, 12, 2766. https://doi.org/10.3390/cancers12102766
Mohanty S, Jyotsana N, Sharma A, Kloos A, Gabdoulline R, Othman B, Lai CK, Schottmann R, Mandhania M, Schmoellerl J, et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia. Cancers. 2020; 12(10):2766. https://doi.org/10.3390/cancers12102766
Chicago/Turabian StyleMohanty, Sagarajit, Nidhi Jyotsana, Amit Sharma, Arnold Kloos, Razif Gabdoulline, Basem Othman, Courteney K. Lai, Renate Schottmann, Madhvi Mandhania, Johannes Schmoellerl, and et al. 2020. "Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia" Cancers 12, no. 10: 2766. https://doi.org/10.3390/cancers12102766
APA StyleMohanty, S., Jyotsana, N., Sharma, A., Kloos, A., Gabdoulline, R., Othman, B., Lai, C. K., Schottmann, R., Mandhania, M., Schmoellerl, J., Grebien, F., Ramsay, E., Thomas, A., Vornlocher, H.-P., Ganser, A., Thol, F., & Heuser, M. (2020). Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia. Cancers, 12(10), 2766. https://doi.org/10.3390/cancers12102766