Mutation Status and Epithelial Differentiation Stratify Recurrence Risk in Chordoid Meningioma—A Multicenter Study with High Prognostic Relevance

Abstract
1. Introduction
2. Results
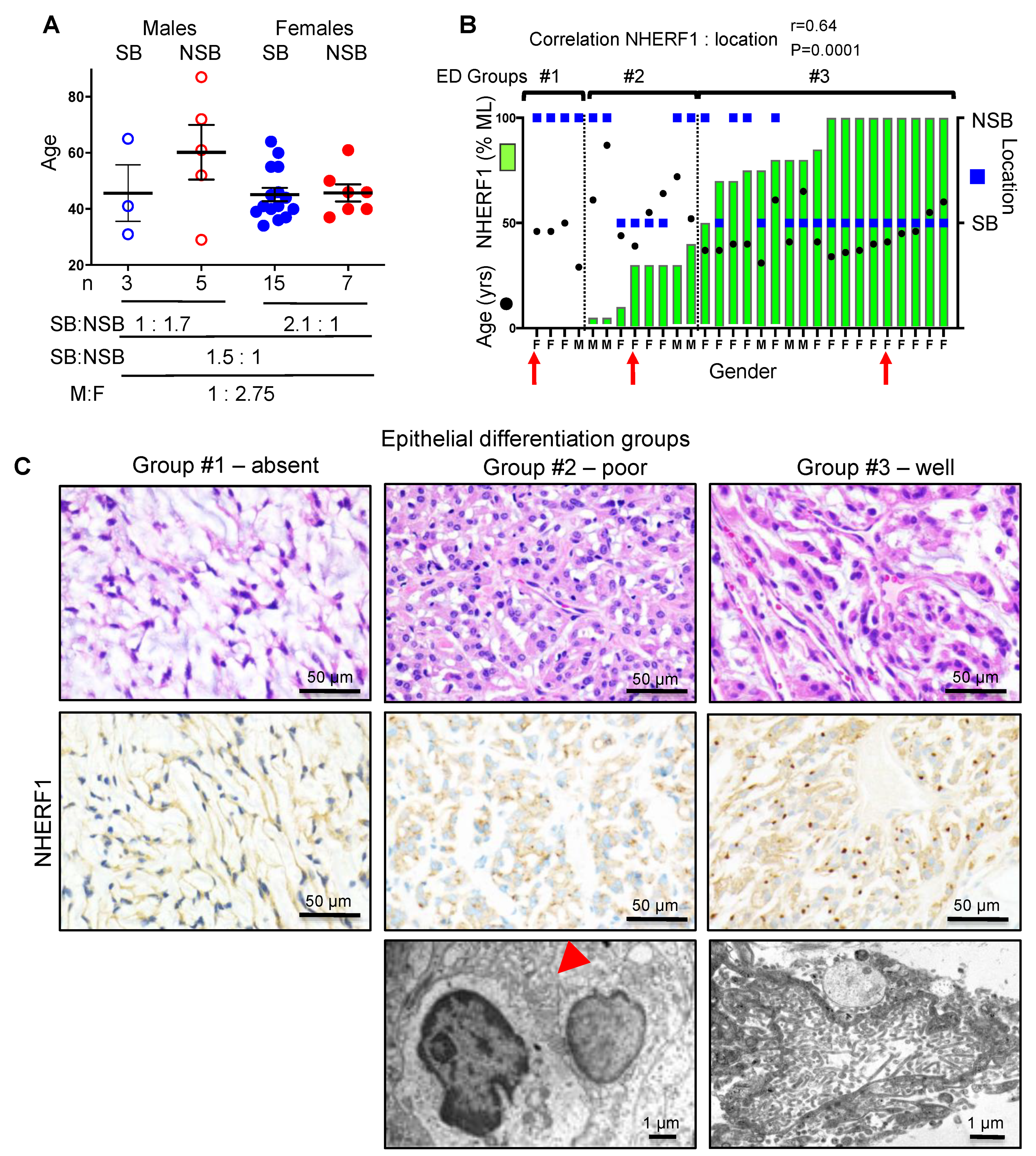
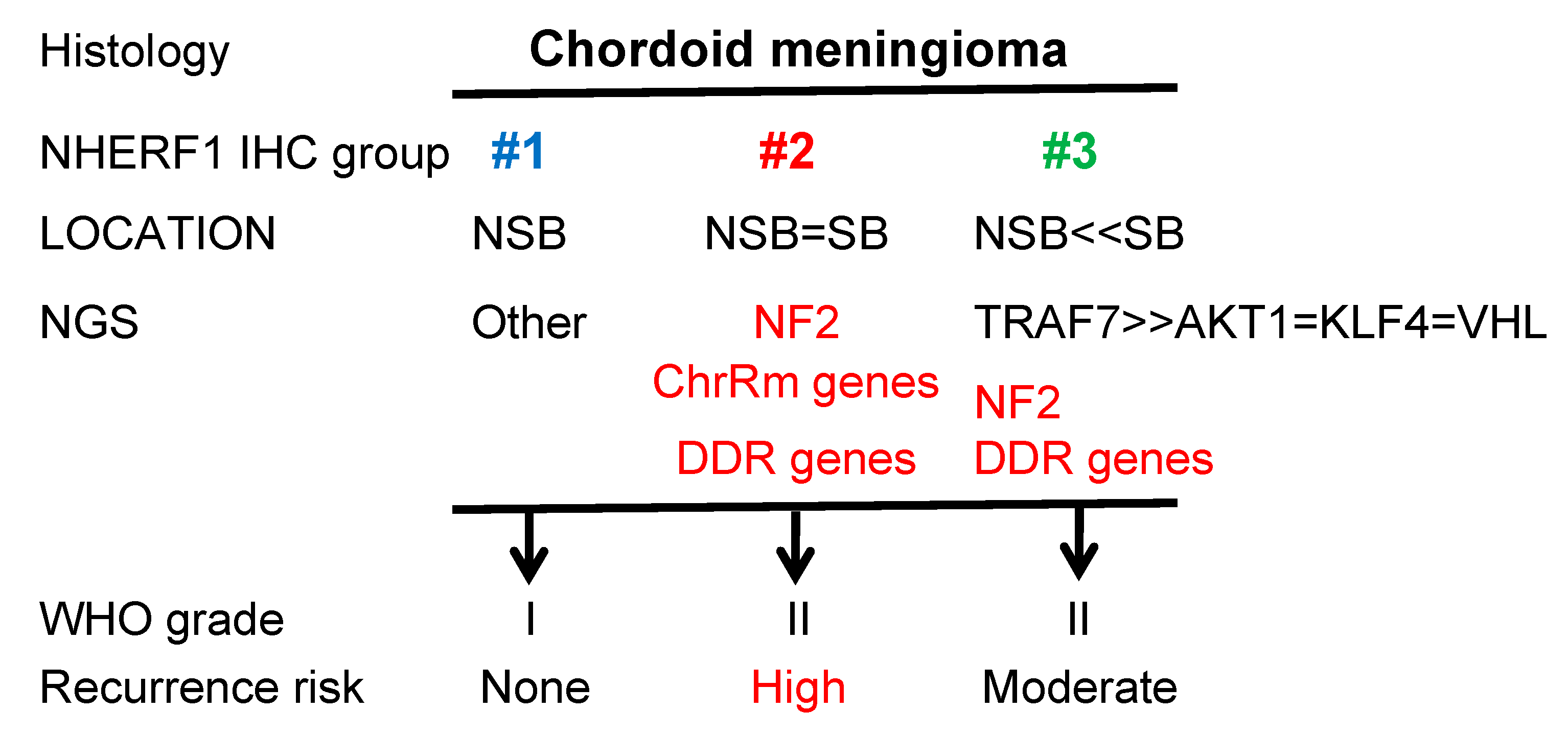
2.1. NHERF1 IHC Defines Three Chordoid Meningioma ED Groups that Correlate with Tumor Location
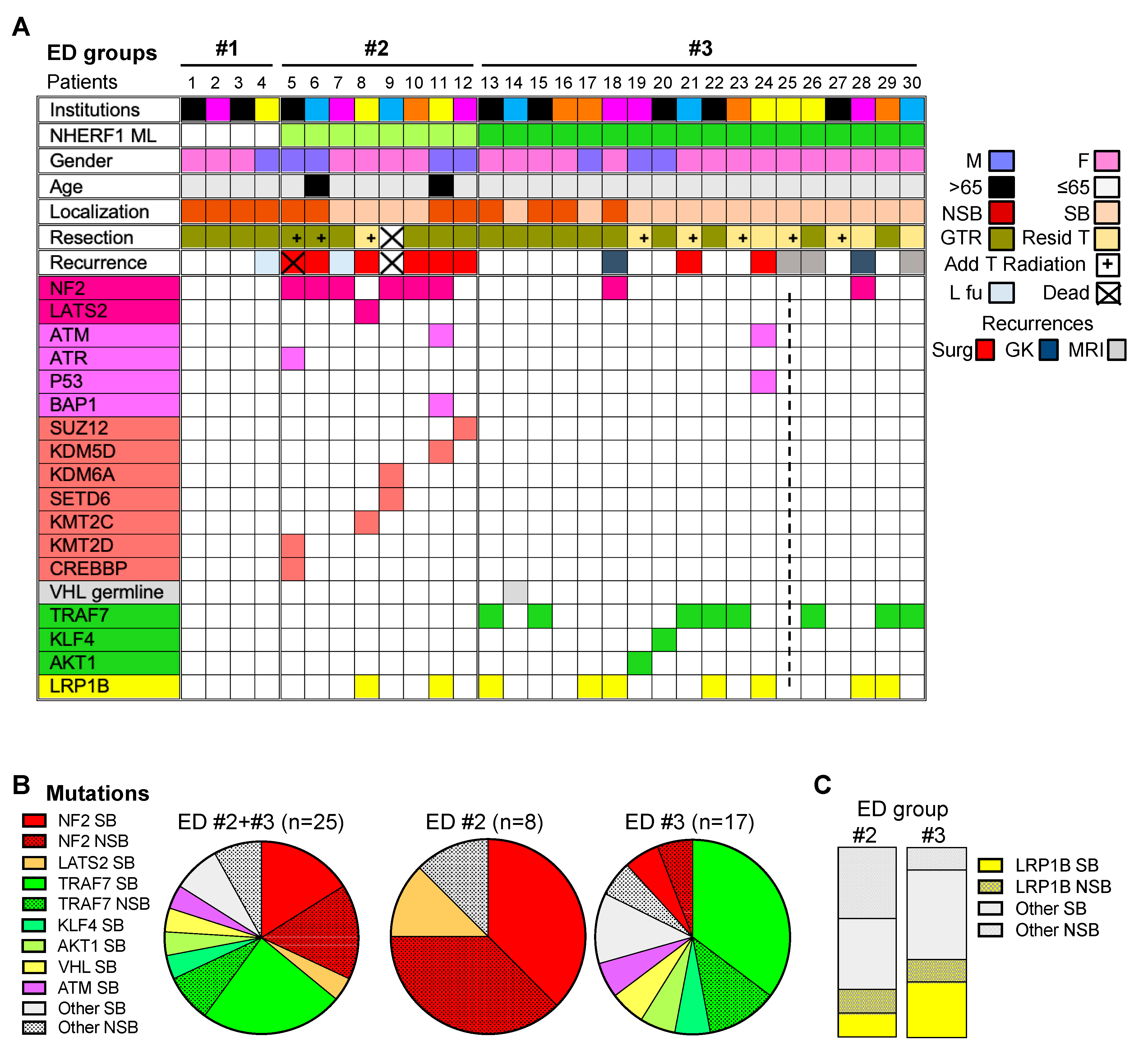
2.2. Chordoid Meningioma ED Groups Exhibit Distinct Gene Mutation Profiles
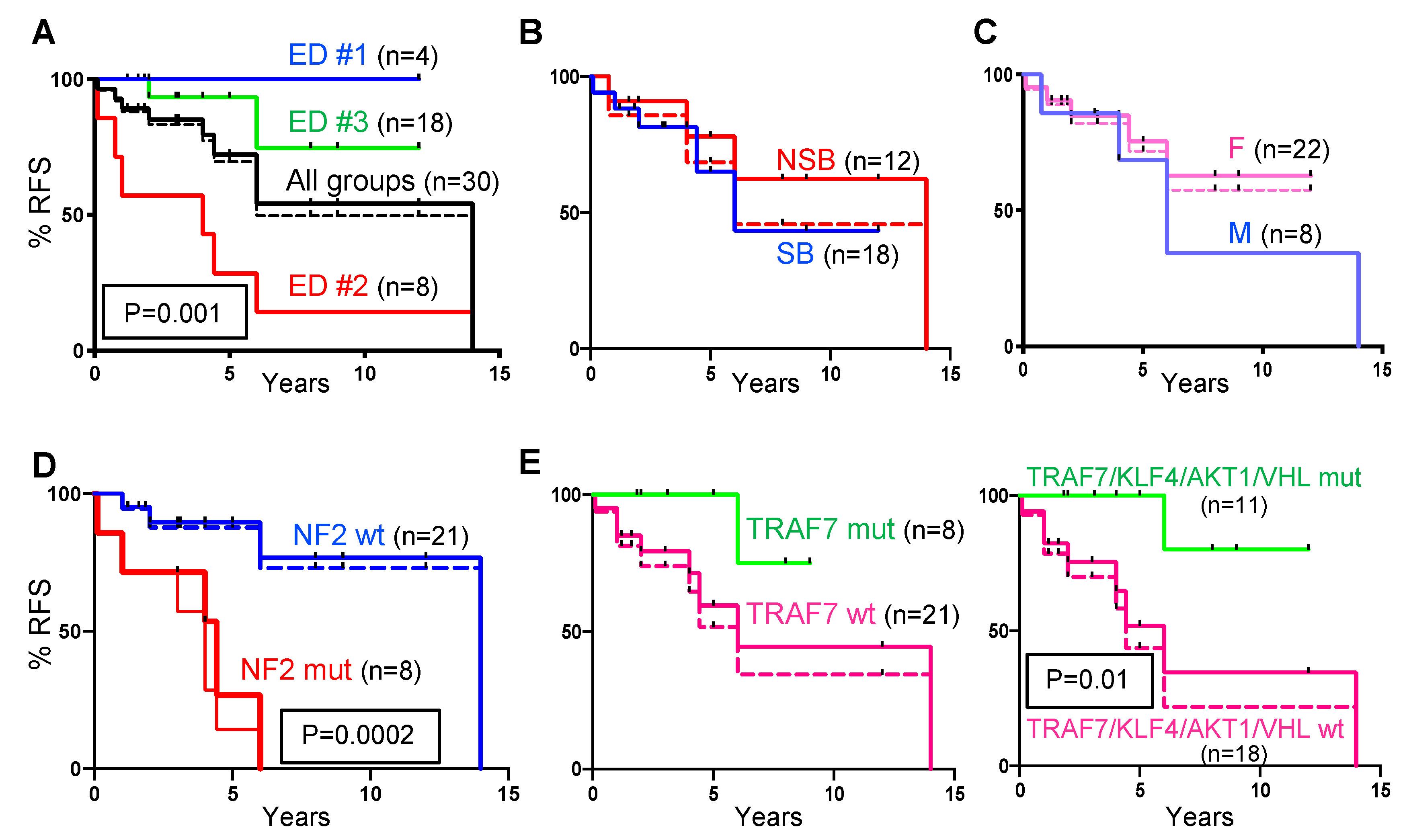
2.3. Recurrence-Free Survival (RFS) Correlates with ED Groups and Mutation Profiles.
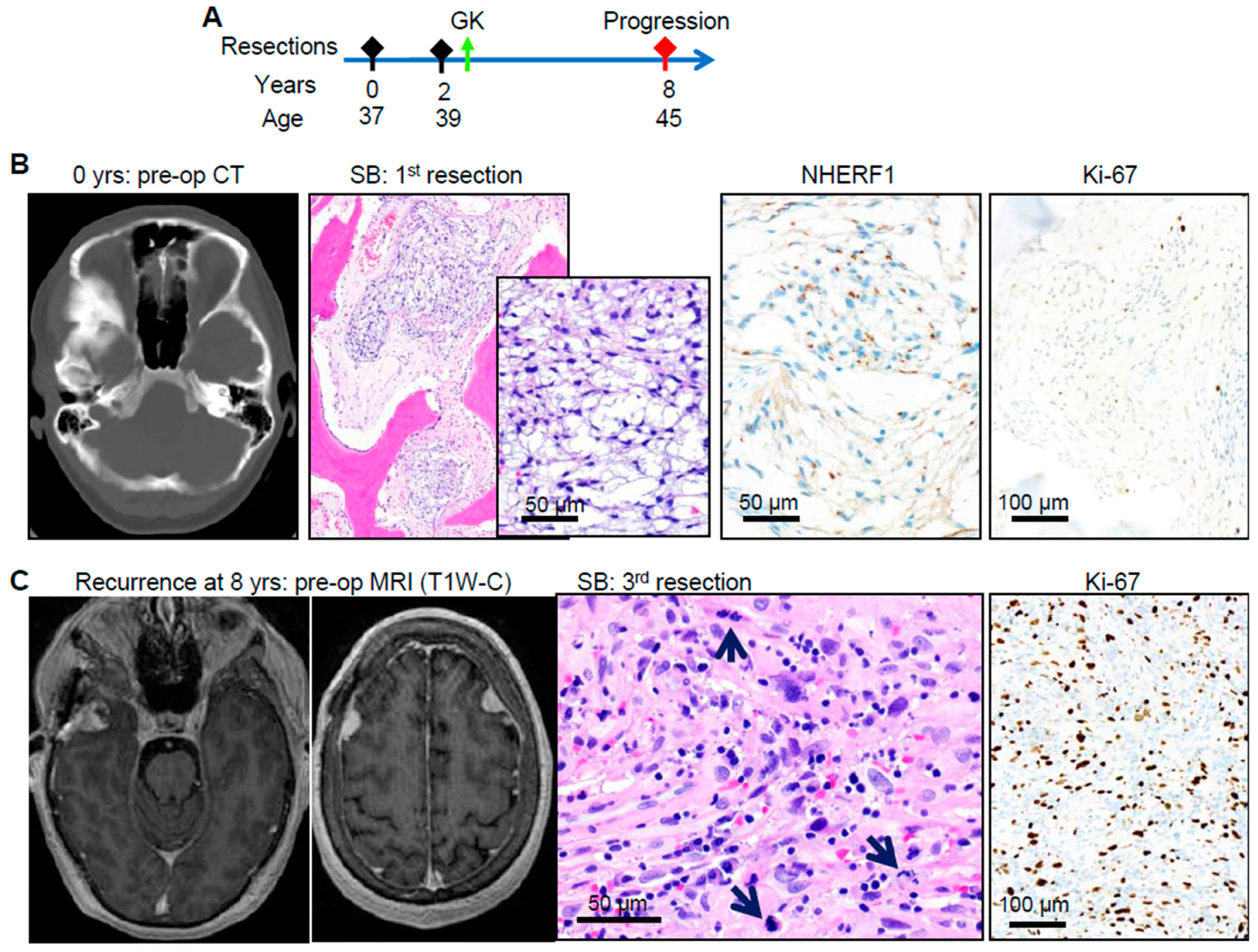
2.4. DDR Germline or Somatic Mutations Induce Aggressive Tumor Phenotypes in Chordoid Meningioma
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Caveneee, W.K. (Eds.) WHO Classification of Tumors of the Central Nervous System; IARC: Lyon, France, 2016. [Google Scholar]
- Goutagny, S.; Nault, J.C.; Mallet, M.; Henin, D.; Rossi, J.Z.; Kalamarides, M. High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain Pathol. 2014, 24, 184–189. [Google Scholar] [CrossRef]
- Sahm, F.; Schrimpf, D.; Olar, A.; Koelsche, C.; Reuss, D.; Bissel, J.; Kratz, A.; Capper, D.; Schefzyk, S.; Hielscher, T.; et al. TERT Promoter Mutations and Risk of Recurrence in Meningioma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef]
- Clark, V.E.; Harmanci, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.E.; Piro, R.M.; Jones, D.T.; Simon, M.; Ketter, R.; Kool, M.; Becker, A.; Sahm, F.; Pusch, S.; Meyer, J.; et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013, 125, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Abedalthagafi, M.; Vaubel, R.A.; Merrill, P.H.; Nayyar, N.; Gill, C.M.; Brewster, R.; Bi, W.L.; Agarwalla, P.K.; Thorner, A.R.; et al. Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro Oncol. 2017, 19, 535–545. [Google Scholar] [CrossRef]
- Smith, M.J.; O’Sullivan, J.; Bhaskar, S.S.; Hadfield, K.D.; Poke, G.; Caird, J.; Sharif, S.; Eccles, D.; Fitzpatrick, D.; Rawluk, D.; et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat. Genet. 2013, 45, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Ruttledge, M.H.; Sarrazin, J.; Rangaratnam, S.; Phelan, C.M.; Twist, E.; Merel, P.; Delattre, O.; Thomas, G.; Nordenskjold, M.; Collins, V.P.; et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat. Genet. 1994, 6, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Kros, J.; de Greve, K.; van Tilborg, A.; Hop, W.; Pieterman, H.; Avezaat, C.; Lekanne Dit Deprez, R.; Zwarthoff, E. NF2 status of meningiomas is associated with tumour localization and histology. J. Pathol. 2001, 194, 367–372. [Google Scholar] [CrossRef]
- Kerr, K.; Qualmann, K.; Esquenazi, Y.; Hagan, J.; Kim, D.H. Familial Syndromes Involving Meningiomas Provide Mechanistic Insight Into Sporadic Disease. Neurosurgery 2018. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M.; Olar, A.; Mobley, B.C.; Faust, P.L.; Raisanen, J.M. Epithelial differentiation with microlumen formation in meningioma: Diagnostic utility of NHERF1/EBP50 immunohistochemistry. Oncotarget 2018, 9, 28652–28665. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morales, F.C.; Takahashi, Y.; Kreimann, E.L.; Georgescu, M.M. Ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc. Natl. Acad. Sci. USA 2004, 101, 17705–17710. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Berryman, M.; Bretscher, A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J. Cell Biol. 1997, 139, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M.; Yell, P.; Mobley, B.C.; Shang, P.; Georgescu, T.; Wang, S.H.; Canoll, P.; Hatanpaa, K.J.; White, C.L., 3rd; Raisanen, J.M. NHERF1/EBP50 is an organizer of polarity structures and a diagnostic marker in ependymoma. Acta Neuropathol. Commun. 2015, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.E.; Aker, F.V.; Scheithauer, B.W. Chordoid meningioma: A clinicopathologic study of 42 cases. Am. J. Surg. Pathol. 2000, 24, 899–905. [Google Scholar] [CrossRef]
- Tang, M.; Wei, H.; Han, L.; Deng, J.; Wang, Y.; Yang, M.; Tang, Y.; Guo, G.; Zhou, L.; Tong, A. Whole-genome sequencing identifies new genetic alterations in meningiomas. Oncotarget 2017, 8, 17070–17080. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic landscape of high-grade meningiomas. NPJ Genom. Med. 2017, 2. [Google Scholar] [CrossRef]
- Beer, A.G.; Zenzmaier, C.; Schreinlechner, M.; Haas, J.; Dietrich, M.F.; Herz, J.; Marschang, P. Expression of a recombinant full-length LRP1B receptor in human non-small cell lung cancer cells confirms the postulated growth-suppressing function of this large LDL receptor family member. Oncotarget 2016, 7, 68721–68733. [Google Scholar] [CrossRef]
- Langbein, S.; Szakacs, O.; Wilhelm, M.; Sukosd, F.; Weber, S.; Jauch, A.; Lopez Beltran, A.; Alken, P.; Kalble, T.; Kovacs, G. Alteration of the LRP1B gene region is associated with high grade of urothelial cancer. Lab. Investig. 2002, 82, 639–643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sonoda, I.; Imoto, I.; Inoue, J.; Shibata, T.; Shimada, Y.; Chin, K.; Imamura, M.; Amagasa, T.; Gray, J.W.; Hirohashi, S.; et al. Frequent silencing of low density lipoprotein receptor-related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res. 2004, 64, 3741–3747. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Taylor-Weiner, A.; Lelic, N.; Jones, R.T.; Kim, J.C.; Francis, J.M.; Abedalthagafi, M.; Borges, L.F.; Coumans, J.V.; Curry, W.T.; et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol. Commun. 2014, 2, 167. [Google Scholar] [CrossRef]
- Wolff, R.K.; Hoffman, M.D.; Wolff, E.C.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.S.; Slattery, M.L. Mutation analysis of adenomas and carcinomas of the colon: Early and late drivers. Genes Chromosomes Cancer 2018, 57, 366–376. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Savardekar, A.R.; Patra, D.P.; Bir, S.; Thakur, J.D.; Mohammed, N.; Bollam, P.; Georgescu, M.M.; Nanda, A. Differential Tumor Progression Patterns in Skull Base Versus Non-Skull Base Meningiomas: A Critical Analysis from a Long-Term Follow-Up Study and Review of Literature. World Neurosurg. 2018, 112, e74–e83. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, Z.; Zhang, W.; Gu, M.; Zhou, Z.D.; Li, B.; Li, J.; Tan, E.K.; Zeng, L. LRRK2 interacts with ATM and regulates Mdm2-p53 cell proliferation axis in response to genotoxic stress. Hum. Mol. Genet. 2017, 26, 4494–4505. [Google Scholar] [CrossRef]
- D’Amico, R.S.; Praver, M.; Zanazzi, G.J.; Englander, Z.K.; Sims, J.S.; Samanamud, J.L.; Ogden, A.T.; McCormick, P.C.; Feldstein, N.A.; McKhann, G.M., 2nd; et al. Subependymomas Are Low-Grade Heterogeneous Glial Neoplasms Defined by Subventricular Zone Lineage Markers. World Neurosurg. 2017, 107, 451–463. [Google Scholar] [CrossRef]
- Murthy, A.; Gonzalez-Agosti, C.; Cordero, E.; Pinney, D.; Candia, C.; Solomon, F.; Gusella, J.; Ramesh, V. NHE-RF, a regulatory cofactor for Na(+)-H+ exchange, is a common interactor for merlin and ERM (MERM) proteins. J. Biol. Chem. 1998, 273, 1273–1276. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Mobley, B.C.; Orr, B.A.; Shang, P.; Lehman, N.L.; Zhu, X.; O’Neill, T.J.; Rajaram, V.; Hatanpaa, K.J.; Timmons, C.F.; et al. NHERF1/EBP50 and NF2 as diagnostic markers for choroid plexus tumors. Acta Neuropathol. Commun. 2016, 4, 55. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Gagea, M.; Cote, G. NHERF1/EBP50 Suppresses Wnt-beta-Catenin Pathway-Driven Intestinal Neoplasia. Neoplasia 2016, 18, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Molina, J.R.; Hamilton, S.R.; Georgescu, M.M. NHERF1/EBP50 is a new marker in colorectal cancer. Neoplasia 2010, 12, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Kreimann, E.L.; Morales, F.C.; de Orbeta-Cruz, J.; Takahashi, Y.; Adams, H.; Liu, T.J.; McCrea, P.D.; Georgescu, M.M. Cortical stabilization of beta-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene 2007, 26, 5290–5299. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Morales, F.C.; Hayashi, Y.; Aldape, K.D.; Georgescu, M.M. Loss of PTEN binding adapter protein NHERF1 from plasma membrane in glioblastoma contributes to PTEN inactivation. Cancer Res. 2010, 70, 6697–6703. [Google Scholar] [CrossRef]
- Chiba, H.; Sakai, N.; Murata, M.; Osanai, M.; Ninomiya, T.; Kojima, T.; Sawada, N. The nuclear receptor hepatocyte nuclear factor 4alpha acts as a morphogen to induce the formation of microvilli. J. Cell Biol. 2006, 175, 971–980. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Cote, G.; Agarwal, N.K.; White, C.L., 3rd. NHERF1/EBP50 controls morphogenesis of 3D colonic glands by stabilizing PTEN and ezrin-radixin-moesin proteins at the apical membrane. Neoplasia 2014, 16, 365–374.e2. [Google Scholar] [CrossRef]
- Louis, D.N.; Ramesh, V.; Gusella, J.F. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol. 1995, 5, 163–172. [Google Scholar] [CrossRef]
- Wellenreuther, R.; Kraus, J.A.; Lenartz, D.; Menon, A.G.; Schramm, J.; Louis, D.N.; Ramesh, V.; Gusella, J.F.; Wiestler, O.D.; von Deimling, A. Analysis of the neurofibromatosis 2 gene reveals molecular variants of meningioma. Am. J. Pathol. 1995, 146, 827–832. [Google Scholar]
- James, M.F.; Lelke, J.M.; Maccollin, M.; Plotkin, S.R.; Stemmer-Rachamimov, A.O.; Ramesh, V.; Gusella, J.F. Modeling NF2 with human arachnoidal and meningioma cell culture systems: NF2 silencing reflects the benign character of tumor growth. Neurobiol. Dis. 2008, 29, 278–292. [Google Scholar] [CrossRef]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet. J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Malmer, B.S.; Feychting, M.; Lonn, S.; Lindstrom, S.; Gronberg, H.; Ahlbom, A.; Schwartzbaum, J.; Auvinen, A.; Collatz-Christensen, H.; Johansen, C.; et al. Genetic variation in p53 and ATM haplotypes and risk of glioma and meningioma. J. Neurooncol. 2007, 82, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Grukova, A.A.; Nikolsky, N.N.; Burova, E.B. Tetraploidization or autophagy: The ultimate fate of senescent human endometrial stem cells under ATM or p53 inhibition. Cell Cycle 2016, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Syljuasen, R.G. Checkpoint adaptation in human cells. Oncogene 2007, 26, 5833–5839. [Google Scholar] [CrossRef] [PubMed]
- Perry, A. Meningiomas. In Russell & Rubinstein’s Pathology of Tumors of the Nervous System, 7th ed.; McLendon, R.E., Rosenblum, M.K., Bigner, D.B., Eds.; Oxford University Press Inc.: New York, NY, USA, 2006; pp. 427–472. [Google Scholar]
- Georgescu, M.M.; Li, Y.; Islam, M.Z.; Notarianni, C.; Sun, H.; Olar, A.; Fuller, G.N. Mutations of the MAPK/TSC/mTOR pathway characterize periventricular glioblastoma with epithelioid SEGA-like morphology-morphological and therapeutic implications. Oncotarget 2019, 10, 4038–4052. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.K.; Zhu, X.; Gagea, M.; White, C.L., 3rd; Cote, G.; Georgescu, M.M. PHLPP2 suppresses the NF-kappaB pathway by inactivating IKKbeta kinase. Oncotarget 2014, 5, 815–823. [Google Scholar] [CrossRef][Green Version]
- Molina, J.R.; Agarwal, N.K.; Morales, F.C.; Hayashi, Y.; Aldape, K.D.; Cote, G.; Georgescu, M.M. PTEN, NHERF1 and PHLPP form a tumor suppressor network that is disabled in glioblastoma. Oncogene 2012, 31, 1264–1274. [Google Scholar] [CrossRef]
- Karsy, M.; Azab, M.A.; Abou-Al-Shaar, H.; Guan, J.; Eli, I.; Jensen, R.L.; Ormond, D.R. Clinical potential of meningioma genomic insights: A practical review for neurosurgeons. Neurosurg. Focus 2018, 44, E10. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ED Groups | ED #1 n = 4; 13.3% | ED #2 n = 8; 26.7% | ED #3 n = 18; 60% | ED #2 + #3 n = 26; 86.7% | Total n = 30; 100% |
|---|---|---|---|---|---|
| Age (median) | 46 | 58 | 40.5 | 42.5 | 44.5 |
| Gender Male:Female | 1:3 | 1:1 | 1:5 | 1:2.7 | 1:2.75 |
| Location SB | 0% | 50% | 78% | 69.2% | 60% |
| Laterality Left:Right:Midline | 1:1:0 | 5:3:0 | 5:3:1 | 7.5:4.5:1 | 8.5:5.5:1 |
| Gross total resection | 100% | 85.7% | 55.5% | 64% | 69% |
| Radiation post-surgery | 0% | 43% | 28% | 32% | 27.6% |
| 1 Surgical recurrence/death | 0% | 100% | 11.1% | 36% | 32% |
| 2 Additional MRI recurrence | 0% | - | 27.8% | 20% | 18% |
| 3 Cumulated recurrence/death | 0% | 100% | 38.9% | 56% | 50% |
| NHERF1 ML extent (average) | 0% | 22.5% | 87% | 67.1% | 58% |
| 4 Chr 22q12.2 (NF2) CN loss NF2 | 25% (n = 4) 0% | 71.5% (n = 7) 75% | 85.7% (n = 7) 11.8% | 78.6% (n = 14) 32% | 66.7% (n = 18) 27.6% |
| Chromatin Remodeling genes | 0% | 62.5% | 0% | 20% | 17.2% |
| DNA damage response genes | 0% | 25% | 5.9% | 12% | 10.3% |
| TRAF7 | 0% | 0% | 47% | 32% | 27.6% |
| 5 KLF4/AKT1/VHL | 0% | 0% | 17.6% | 12% | 10.3% |
| LRP1B | 0% | 25% | 41.2% | 36% | 31% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgescu, M.-M.; Nanda, A.; Li, Y.; Mobley, B.C.; Faust, P.L.; Raisanen, J.M.; Olar, A. Mutation Status and Epithelial Differentiation Stratify Recurrence Risk in Chordoid Meningioma—A Multicenter Study with High Prognostic Relevance. Cancers 2020, 12, 225. https://doi.org/10.3390/cancers12010225
Georgescu M-M, Nanda A, Li Y, Mobley BC, Faust PL, Raisanen JM, Olar A. Mutation Status and Epithelial Differentiation Stratify Recurrence Risk in Chordoid Meningioma—A Multicenter Study with High Prognostic Relevance. Cancers. 2020; 12(1):225. https://doi.org/10.3390/cancers12010225
Chicago/Turabian StyleGeorgescu, Maria-Magdalena, Anil Nanda, Yan Li, Bret C. Mobley, Phyllis L. Faust, Jack M. Raisanen, and Adriana Olar. 2020. "Mutation Status and Epithelial Differentiation Stratify Recurrence Risk in Chordoid Meningioma—A Multicenter Study with High Prognostic Relevance" Cancers 12, no. 1: 225. https://doi.org/10.3390/cancers12010225
APA StyleGeorgescu, M.-M., Nanda, A., Li, Y., Mobley, B. C., Faust, P. L., Raisanen, J. M., & Olar, A. (2020). Mutation Status and Epithelial Differentiation Stratify Recurrence Risk in Chordoid Meningioma—A Multicenter Study with High Prognostic Relevance. Cancers, 12(1), 225. https://doi.org/10.3390/cancers12010225