Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer

 ,
,
Abstract
1. Introduction
2. Results
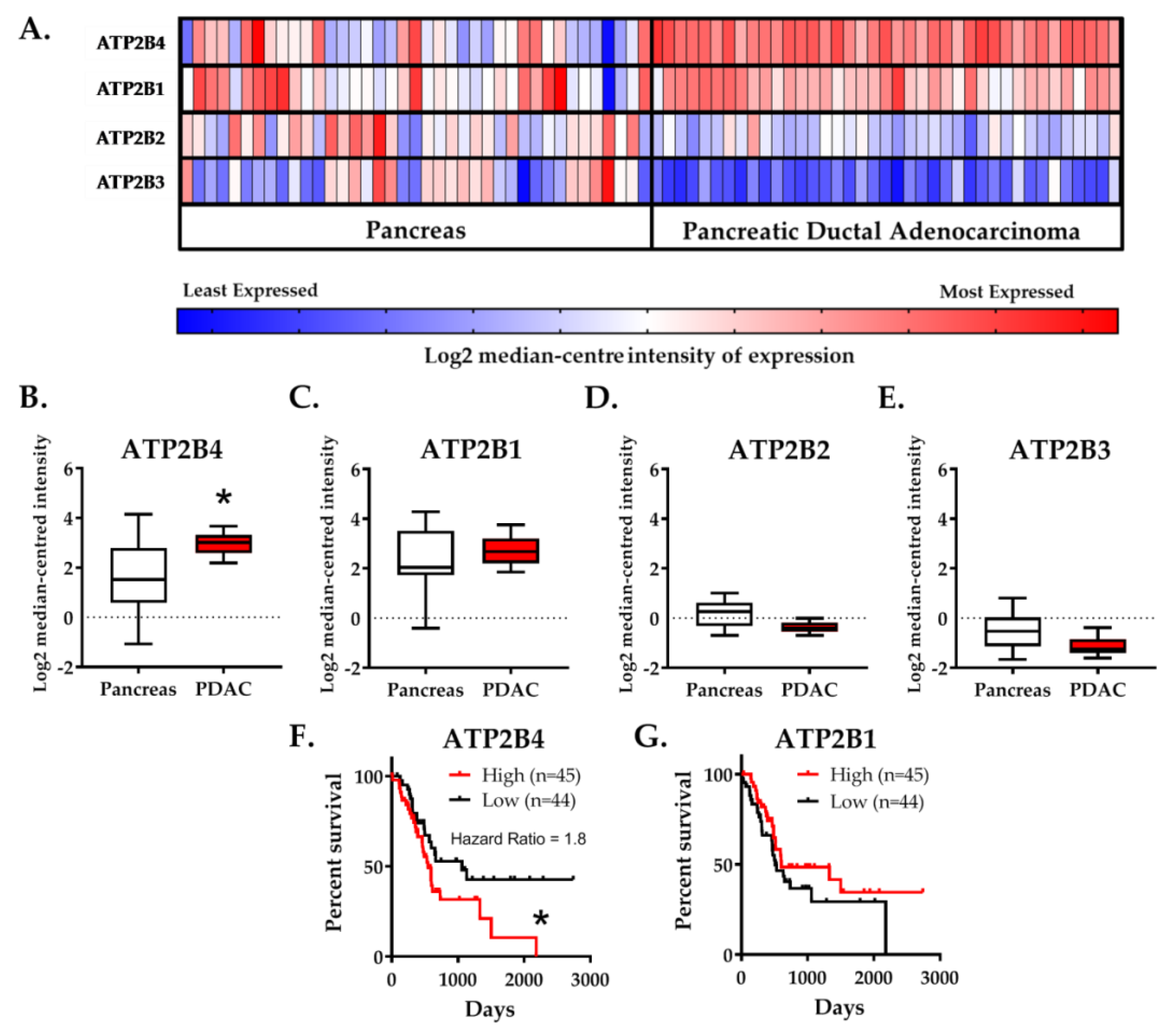
2.1. Expression of the PMCA4 Gene, ATP2B4, Is Correlated with PDAC and Poor Patient Survival
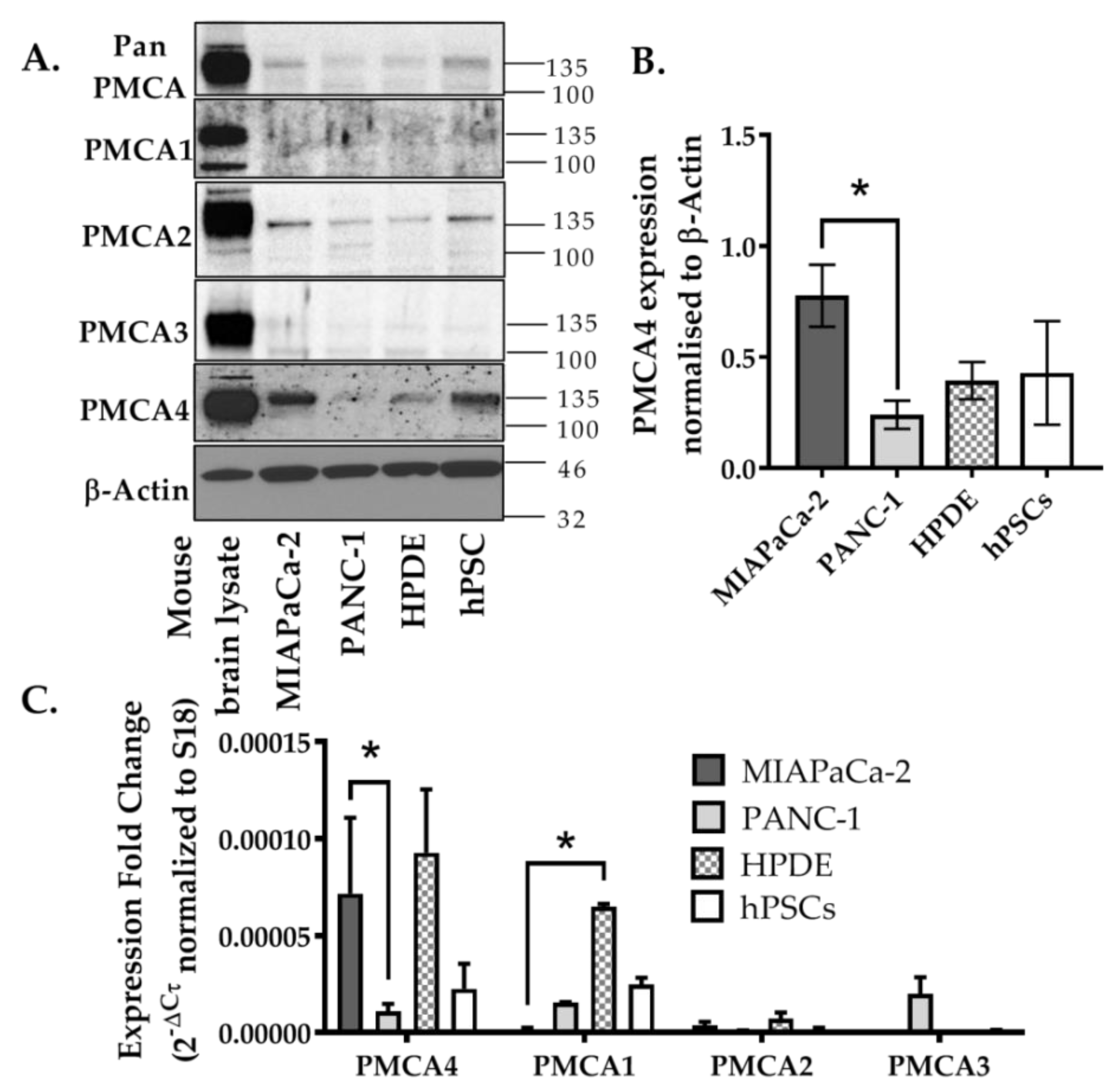
2.2. PMCA4 Is the Major PMCA Isoform Expressed in MIA PaCa-2 Pancreatic Cancer Cell Line
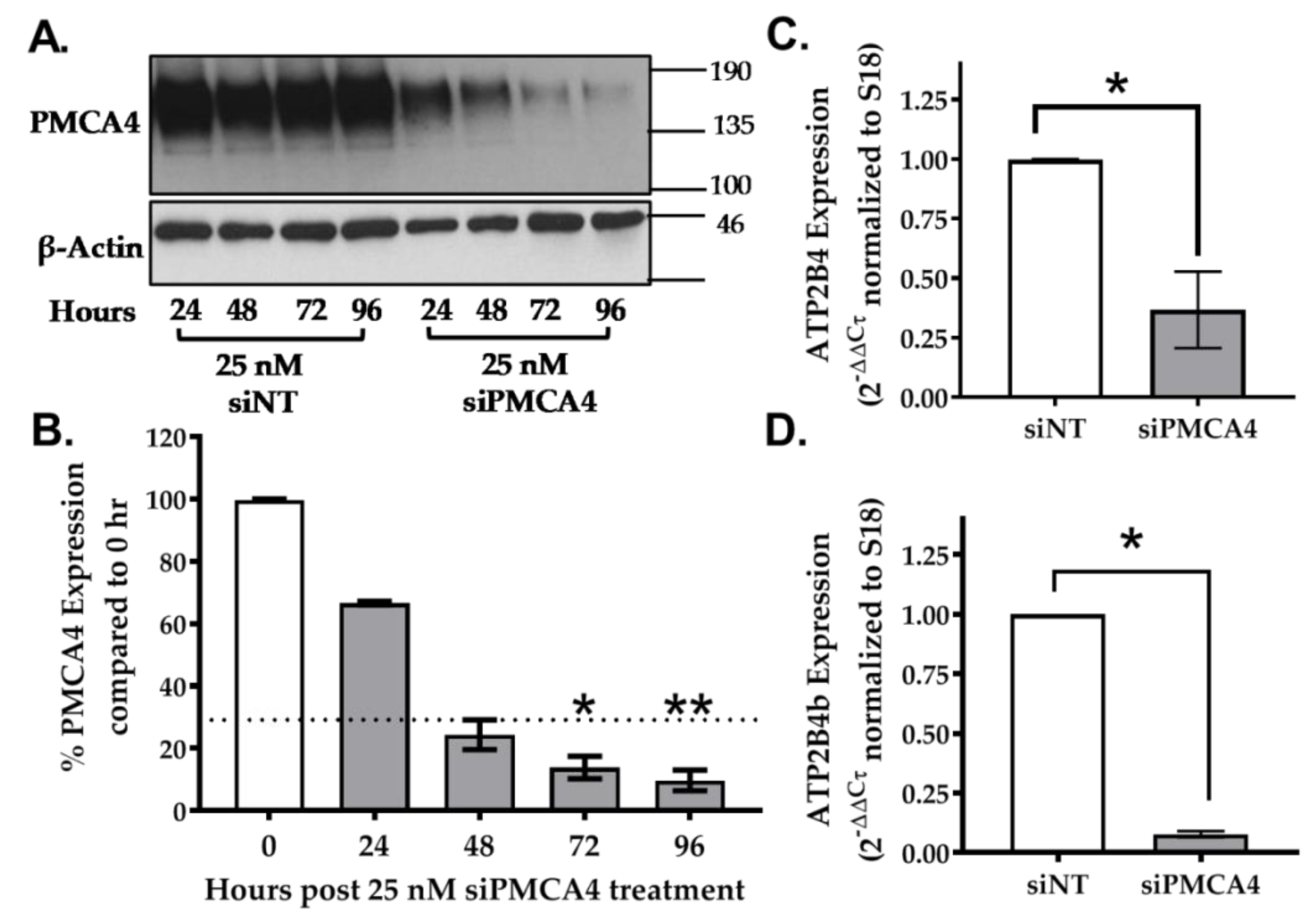
2.3. siRNA Knockdown of ATP2B4 mRNA and PMCA4 Protein Expression in MIA PaCa-2 Cells
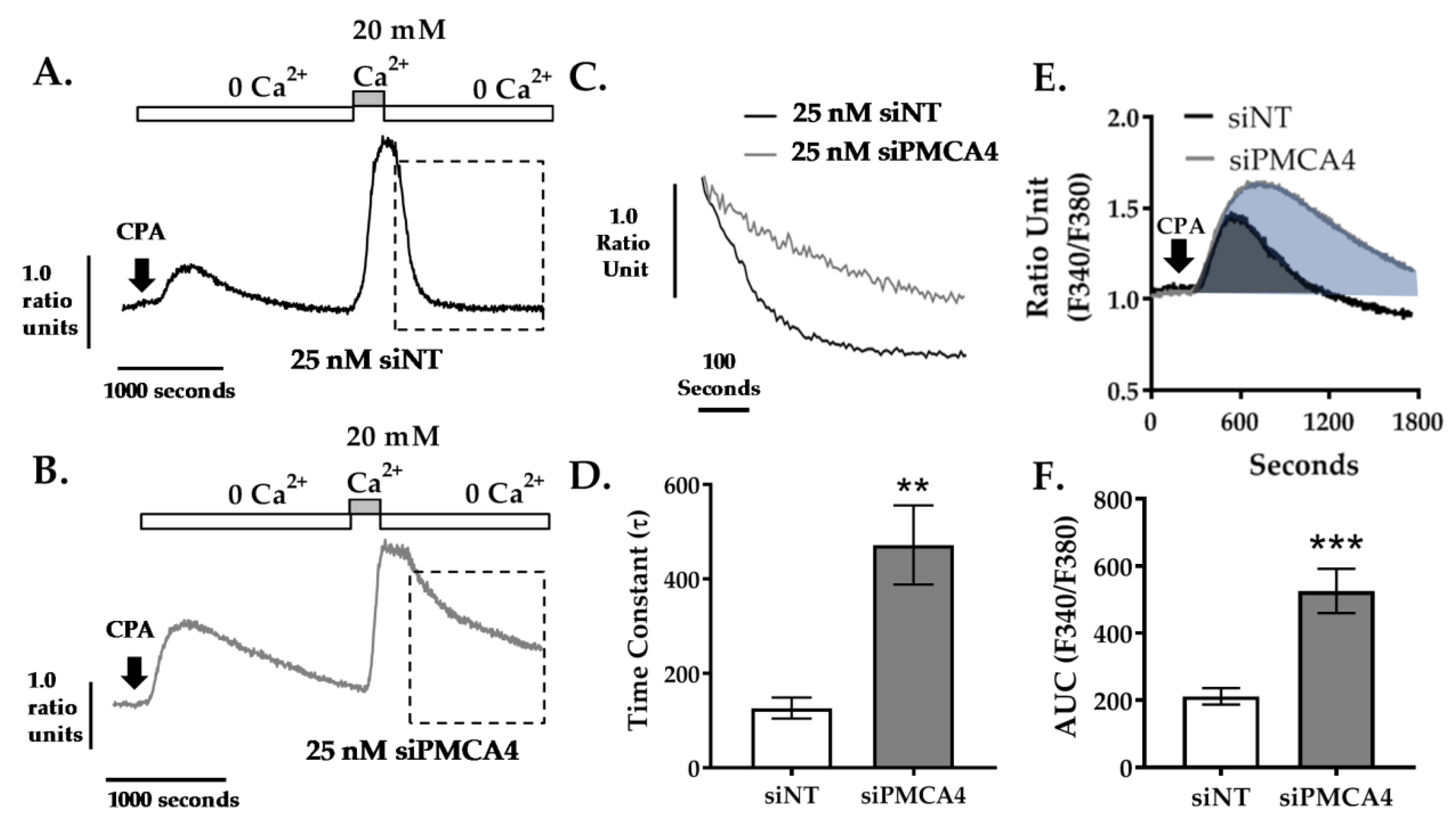
2.4. PMCA4 Is the Major Functional Ca2+ Efflux Pathway in MIA PaCa-2 Cells
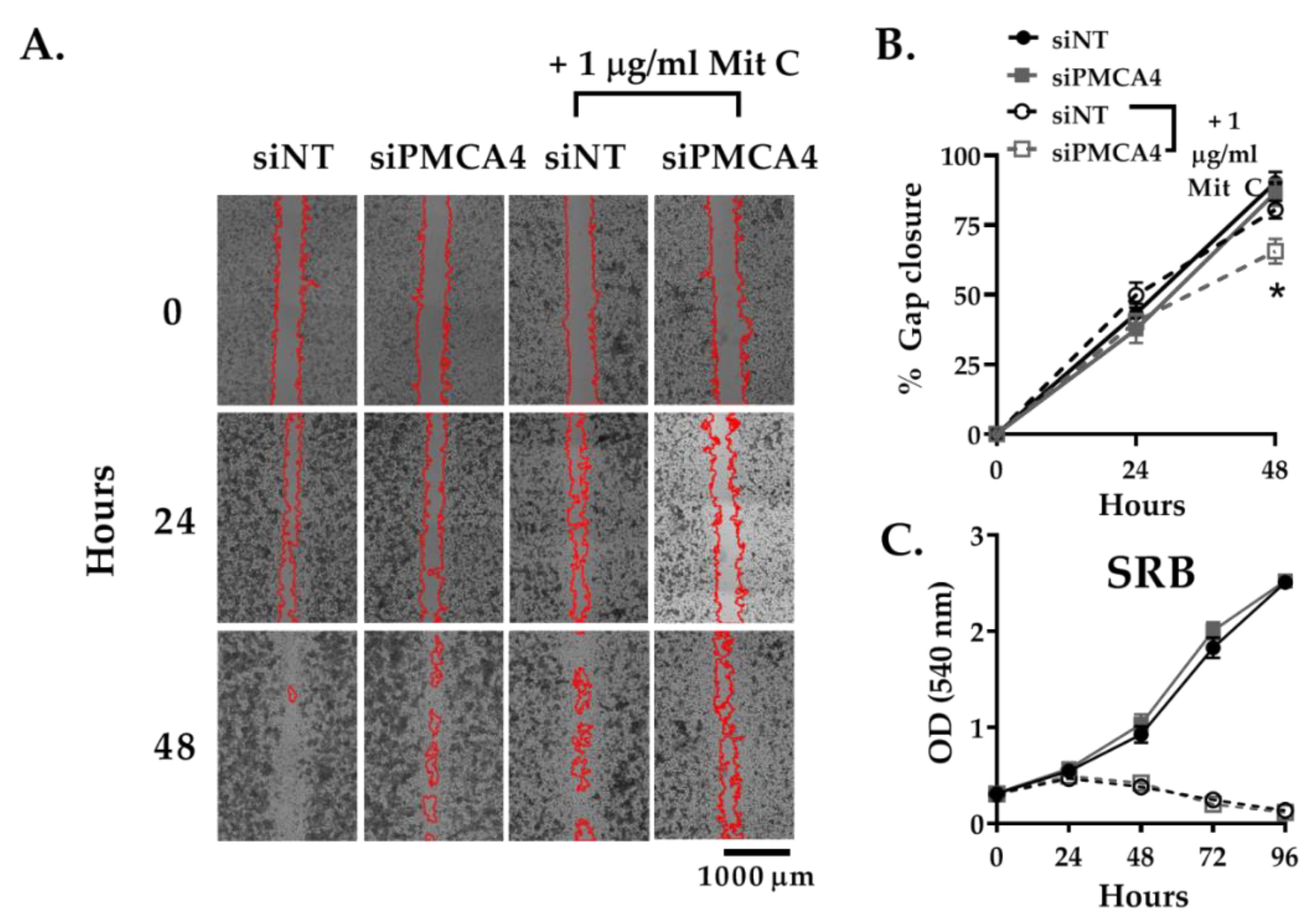
2.5. PMCA4 Knockdown Inhibits Cell Migration Independent of Cell Proliferation
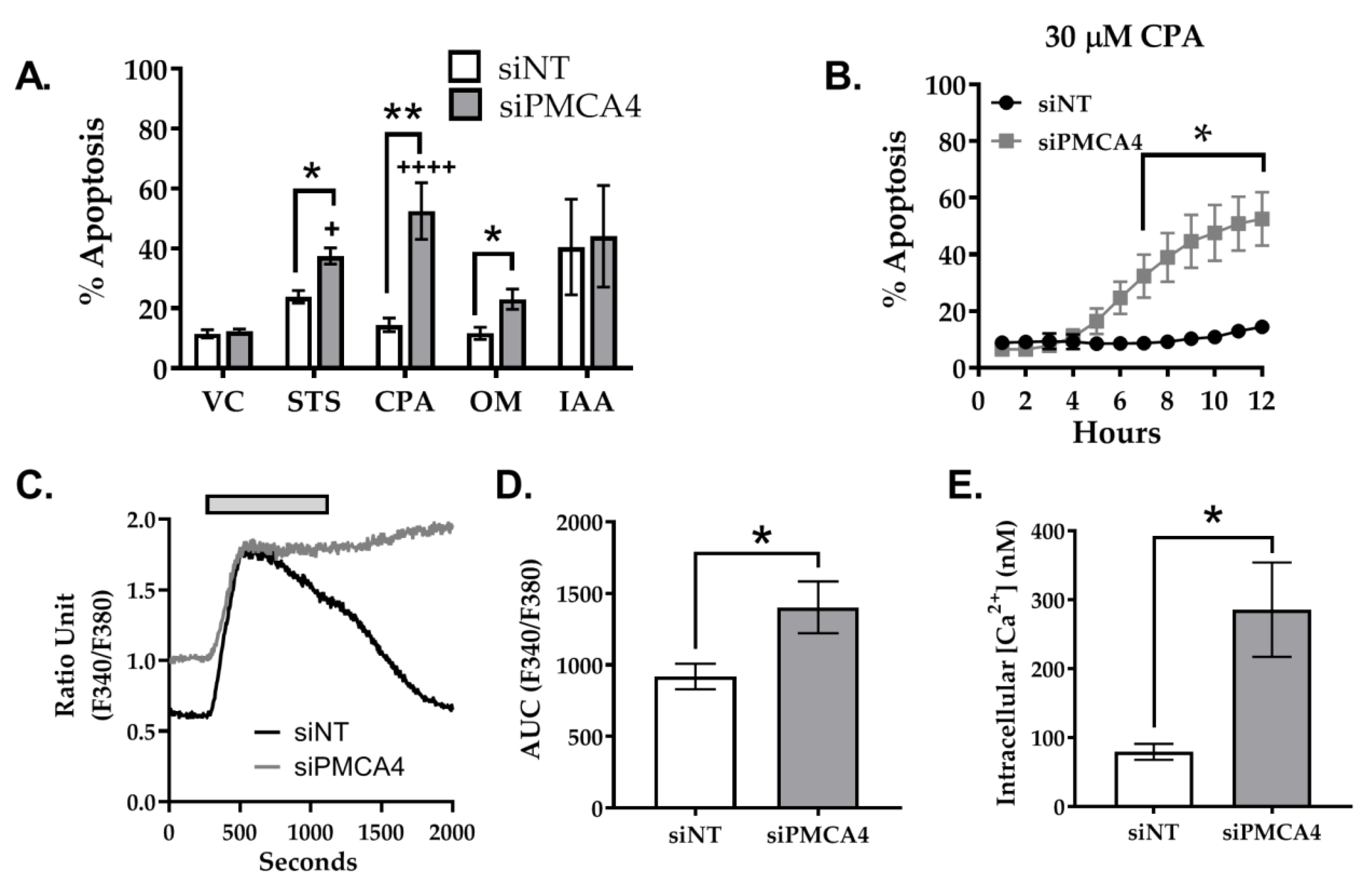
2.6. PMCA4 Knockdown Sensitizes MIA PaCa-2 Cells to Apoptosis
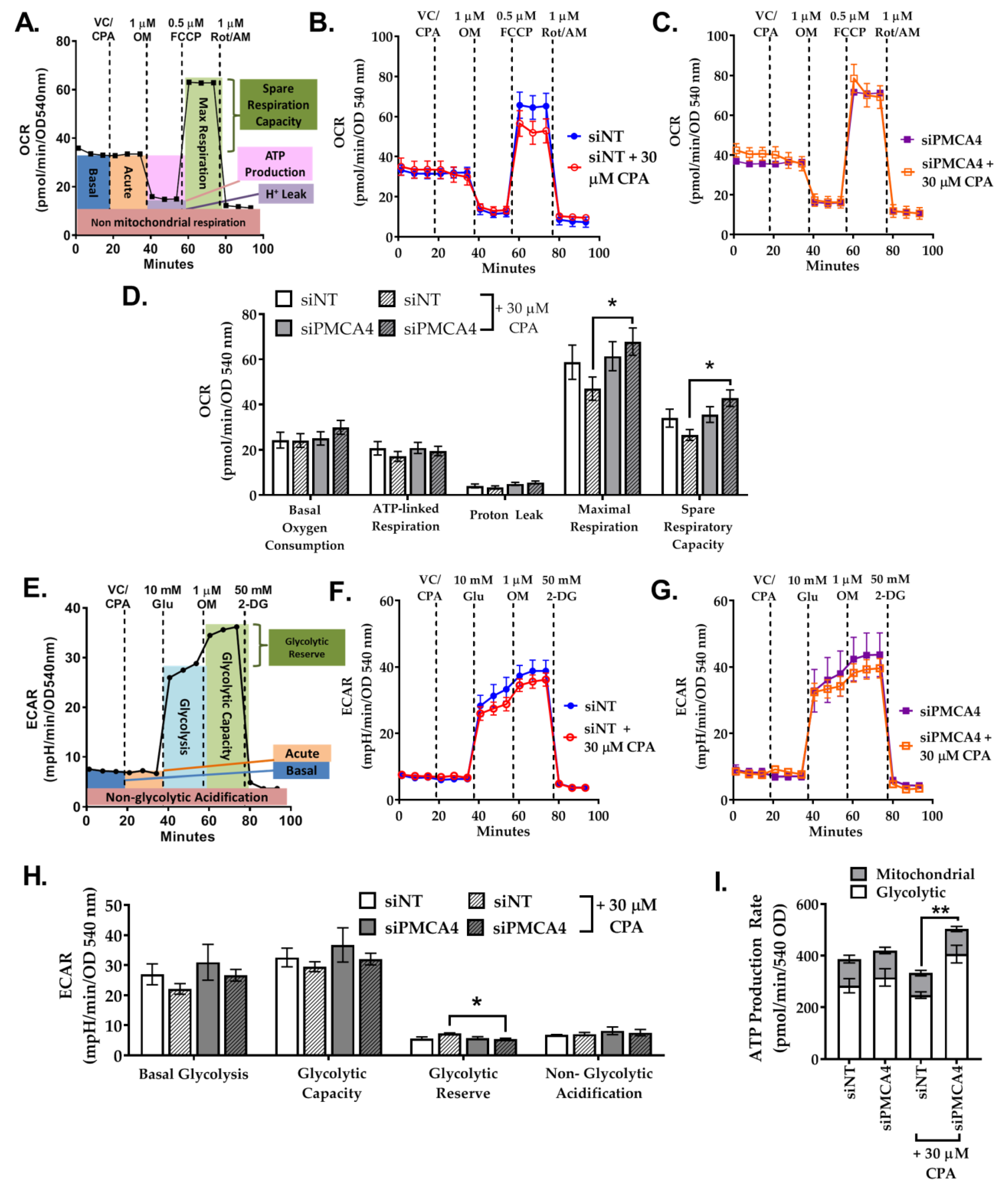
2.7. PMCA4 Knockdown Alters Metabolic Flexibility during Ca2+ Overload
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals and Reagents
4.3. Data-Mining
4.4. SiRNA Knockdown of PMCA4 Expression
4.5. Quantitative RT-qPCR
4.6. Western Immunoblotting
4.7. Sulforhodamine B (SRB) Cell Viability Assay
4.8. PMCA Activity Assay (Ca2+ Clearance)
4.9. Ca2+ Overload Assay
4.10. Cell Migration
4.11. Caspase3/7 Apoptosis Assay
4.12. Seahorse Live-Cell Metabolic Assays
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Office for National Statistics. Cancer Survival in England: Adults Diagnosed in 2009 to 2013, Followed up to 2014; Office for National Statistics: London, UK, 2015; pp. 1–14.
- Le Large, T.Y.S.; Bijlsma, M.F.; Kazemier, G.; van Laarhoven, H.W.M.; Giovannetti, E.; Jimenez, C.R. Key biological processes driving metastatic spread of pancreatic cancer as identified by multi-omics studies. Semin. Cancer Biol. 2017, 44, 153–169. [Google Scholar] [CrossRef]
- Quaresma, M.; Coleman, M.P.; Rachet, B. 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971–2011: A population-based study. Lancet 2015, 385, 1206–1218. [Google Scholar] [CrossRef]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.I.E. Metabolic regulation of the PMCA: Role in cell death and survival. Cell Calcium 2018, 69, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Criddle, D.N.; Gerasimenko, J.V.; Baumgartner, H.K.; Jaffar, M.; Voronina, S.; Sutton, R.; Petersen, O.H.; Gerasimenko, O.V. Calcium signalling and pancreatic cell death: Apoptosis or necrosis? Cell Death Differ. 2007, 14, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- James, A.D.; Patel, W.; Butt, Z.; Adiamah, M.; Dakhel, R.; Latif, A.; Uggenti, C.; Swanton, E.; Imamura, H.; Siriwardena, A.K.; et al. The Plasma Membrane Calcium Pump in Pancreatic Cancer Cells Exhibiting the Warburg Effect Relies on Glycolytic ATP. J. Biol. Chem. 2015, 290, 24760–24771. [Google Scholar] [CrossRef] [PubMed]
- James, A.D.; Chan, A.; Erice, O.; Siriwardena, A.K.; Bruce, J.I.E. Glycolytic ATP fuels the plasma membrane calcium pump critical for pancreatic cancer cell survival. J. Biol. Chem. 2013, 288, 36007–36019. [Google Scholar] [CrossRef]
- Strehler, E.E.; Zacharias, D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [CrossRef]
- Lopreiato, R.; Giacomello, M.; Carafoli, E. The plasma membrane calcium pump: New ways to look at an old enzyme. J. Biol. Chem. 2014, 289, 10261–10268. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.S.; Shin, D.M.; Liu, L.H.; Shull, G.E.; Muallem, S. Plasticity and adaptation of Ca2+ signaling and Ca2+-dependent exocytosis in SERCA2+/- mice. EMBO J. 2001, 20, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- Curry, M.C.; Luk, N.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Distinct regulation of cytoplasmic calcium signals and cell death pathways by different plasma membrane calcium ATPase isoforms in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2012, 287, 28598–28608. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Wang, X.; Han, J. Resistance to tumor necrosis factor-induced cell death mediated by PMCA4 deficiency. Mol. Cell. Biol. 2001, 21, 8276–8288. [Google Scholar] [CrossRef]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjö, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezene, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef]
- Puchulu-Campanella, E.; Chu, H.; Anstee, D.J.; Galan, J.A.; Tao, W.A.; Low, P.S. Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane. J. Biol. Chem. 2013, 288, 848–858. [Google Scholar] [CrossRef]
- Paul, R.J.; Hardin, C.D.; Raeymaekers, L.; Wuytack, F.; Casteels, R. Preferential support of Ca2+ uptake in smooth muscle plasma membrane vesicles by an endogenous glycolytic cascade. FASEB J. 1989, 3, 2298–2301. [Google Scholar] [CrossRef]
- Hardin, C.D. Comparison of endogenous and exogenous sources of ATP in fueling Ca2+ uptake in smooth muscle plasma membrane vesicles. J. Gen. Physiol. 1992, 99, 21–40. [Google Scholar] [CrossRef]
- Kemp, R.G.; Gunasekera, D. Evolution of the allosteric ligand sites of mammalian phosphofructo-1-kinase. Biochemistry 2002, 41, 9426–9430. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Mesecar, A.; Heath, P.J.; Shi, W.; Nowak, T.; Stoddard, B.L. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 1998, 6, 195–210. [Google Scholar] [CrossRef]
- Aung, C.S.; Ye, W.; Plowman, G.; Peters, A.A.; Monteith, G.R.; Roberts-Thomson, S.J. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis 2009, 30, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Furukawa, Y.; Nakagawa, H.; Tsunoda, T.; Ohigashi, H.; Murata, K.; Ishikawa, O.; Ohgaki, K.; Kashimura, N.; Miyamoto, M.; et al. Genome-wide cDNA microarray analysis of gene expression profiles in pancreatic cancers using populations of tumor cells and normal ductal epithelial cells selected for purity by laser microdissection. Oncogene 2004, 23, 2385–2400. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef]
- Furler, R.L.; Nixon, D.F.; Brantner, C.A.; Popratiloff, A.; Uittenbogaart, C.H. TGF-β sustains tumor progression through biochemical and mechanical signal transduction. Cancers 2018, 10, 199. [Google Scholar] [CrossRef]
- Porcelli, L.; Iacobazzi, R.M.; Di Fonte, R.; Serratì, S.; Intini, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-β signaling activation by mast cells contribute to resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers 2019, 11, 330. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pander, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Badea, L.; Herlea, V.; Dima, S.O.; Dumitrascu, T.; Popescu, I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato Gastroenterol. 2008, 55, 2016–2027. [Google Scholar]
- Uhlen, M. Expression of ATP2B4 in Pancreatic Cancer—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000058668-ATP2B4/pathology (accessed on 17 January 2019).
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (PANC-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Yunis, A.A.; Arimura, G.K.; Russin, D.J. Human pancreatic carcinoma (MIA PaCa-2) in continuous culture: Sensitivity to asparaginase. Int. J. Cancer 1977, 19, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.V.; Goto, M.; Magnuson, B.; Abraham, S.; Ramanathan, N.; Hotaling, E.; Alaniz, A.A.; Kumar-Sinha, C.; Dziubinski, M.L.; Urs, S.; et al. HNF1A is a novel oncogene that regulates human pancreatic cancer stem cell properties. eLife 2018, 7, e33947. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Duguid, W.P.; Rosenberg, L.; Viallet, J.; Galloway, D.A.; Tsao, M.S. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am. J. Pathol. 1996, 148, 1763–1770. [Google Scholar] [PubMed]
- Won, J.H.; Zhang, Y.; Ji, B.; Logsdon, C.D.; Yule, D.I. Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol. Biol. Cell 2011, 22, 421–436. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-Associated Stromal Fibroblasts Promote Pancreatic Tumor Progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Lenggenhager, D.; Amrutkar, M.; Sántha, P.; Aasrum, M.; Löhr, J.-M.; Gladhaug, I.P.; Verbeke, C.S. Commonly Used Pancreatic Stellate Cell Cultures Differ Phenotypically and in Their Interactions with Pancreatic Cancer Cells. Cells 2019, 8, 23. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef]
- Afroze, T.; Yang, G.; Khoshbin, A.; Tanwir, M.; Tabish, T.; Momen, A.; Husain, M. Calcium efflux activity of plasma membrane Ca2+ ATPase-4 (PMCA4) mediates cell cycle progression in vascular smooth muscle cells. J. Biol. Chem. 2014, 289, 7221–7231. [Google Scholar] [CrossRef]
- Fu, P.; Thompson, J.A.; Bach, L.A. Promotion of cancer cell migration: An insulin-like growth factor (IGF)-independent action of IGF-binding protein-6. J. Biol. Chem. 2007, 282, 22298–22306. [Google Scholar] [CrossRef]
- Campbell, L.; Emmerson, E.; Davies, F.; Gilliver, S.C.; Krust, A.; Chambon, P.; Ashcroft, G.S.; Hardman, M.J. Estrogen promotes cutaneous wound healing via estrogen receptor β independent of its antiinflammatory activities. J. Exp. Med. 2010, 207, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Aung, C.S.; Kruger, W.A.; Poronnik, P.; Roberts-Thomson, S.J.; Monteith, G.R. Plasma membrane Ca2+-ATPase expression during colon cancer cell line differentiation. Biochem. Biophys. Res. Commun. 2007, 355, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Miani, M.; Barthson, J.; Colli, M.L.; Brozzi, F.; Cnop, M.; Eizirik, D.L. Endoplasmic reticulum stress sensitizes pancreatic beta cells to interleukin-1b-induced apoptosis via Bim/A1 imbalance. Cell Death Dis. 2013, 4, e701. [Google Scholar] [CrossRef] [PubMed]
- Marinho-Carvalho, M.M.; Costa-Mattos, P.V.; Spitz, G.A.; Zancan, P.; Sola-Penna, M. Calmodulin upregulates skeletal muscle 6-phosphofructo-1-kinase reversing the inhibitory effects of allosteric modulators. Biochim. Biophys. Acta Proteins Proteom. 2009, 1794, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Romero, N.; Rogers, G.; Neilson, A.; Dranka, B.P. Quantifying Cellular ATP Production Rate Using Agilent Seahorse XF Technology; White Paper; Agilent Technologies, Inc.: Lexington, MA, USA, 2018. [Google Scholar]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Okunade, G.W.; Miller, M.L.; Pyne, G.J.; Sutliff, R.L.; O’Connor, K.T.; Neumann, J.C.; Andringa, A.; Miller, D.A.; Prasad, V.; Doetschman, T.; et al. Targeted Ablation of Plasma Membrane Ca2+-ATPase (PMCA) 1 and 4 Indicates a Major Housekeeping Function for PMCA1 and a Critical Role in Hyperactivated Sperm Motility and Male Fertility for PMCA4. J. Biol. Chem. 2004, 279, 33742–33750. [Google Scholar] [CrossRef]
- Schuh, K.; Cartwright, E.J.; Jankevics, E.; Bundschu, K.; Liebermann, J.; Williams, J.C.; Armesilla, A.L.; Emerson, M.; Oceandy, D.; Knobeloch, K.-P.; et al. Plasma membrane Ca2+ ATPase 4 is required for sperm motility and male fertility. J. Biol. Chem. 2004, 279, 28220–28226. [Google Scholar] [CrossRef]
- Pande, J.; Grover, A.K. Plasma membrane calcium pumps in smooth muscle: From fictional molecules to novel inhibitors. Can. J. Physiol. Pharmacol. 2005, 83, 743–754. [Google Scholar] [CrossRef]
- Lestari, S.W.; Miati, D.N.; Seoharso, P.; Sugiyanto, R.; Pujianto, D.A. Sperm Na+, K+-ATPase α4 and plasma membrane Ca2+-ATPase (PMCA) 4 regulation in asthenozoospermia. Syst. Biol. Reprod. Med. 2017, 63, 294–302. [Google Scholar] [CrossRef]
- Ritchie, M.F.; Samakai, E.; Soboloff, J. STIM1 is required for attenuation of PMCA-mediated Ca2+ clearance during T-cell activation. EMBO J. 2012, 31, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Frigeri, M.; De Dosso, S.; Castillo-Fernandez, O.; Feuerlein, K.; Neuenschwander, H.; Saletti, P. Chemotherapy in patients with advanced pancreatic cancer: Too close to death? Support. Care Cancer 2013, 21, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Hudson, E.; Reza, S.; Thomas, M.; Crosby, T.; Maughan, T. Pancreatic Cancer within a UK Cancer Network with Special Emphasis on Locally Advanced Non-metastatic Pancreatic Cancer. Clin. Oncol. 2008, 20, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.-C.; Kuo, G.-H.; Chang, S.-W.; Tsai, P.-J. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res. Int. 2015, 2015, 409245. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.-S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef]
- Talarico, E.F. Plasma membrane calcium-ATPase isoform four distribution changes during corneal epithelial wound healing. Mol. Vis. 2010, 16, 2259–2272. [Google Scholar]
- Peters, A.A.; Milevskiy, M.J.G.; Lee, W.C.; Curry, M.C.; Smart, C.E.; Saunus, J.M.; Reid, L.; da Silva, L.; Marcial, D.L.; Dray, E.; et al. The calcium pump plasma membrane Ca2+-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin. Sci. Rep. 2016, 6, 25505. [Google Scholar] [CrossRef]
- Modi, S.; Kir, D.; Banerjee, S.; Saluja, A. Control of Apoptosis in Treatment and Biology of Pancreatic Cancer. J. Cell. Biochem. 2016, 117, 279–288. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.D.S.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2502–2511. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; de Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef]
- Graier, W.F.; Malli, R. Mitochondrial calcium: A crucial hub for cancer cell metabolism? Transl. Cancer Res. 2017, 6, S1124–S1127. [Google Scholar] [CrossRef]
- Sola-Penna, M.; Da Silva, D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Zancan, P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life 2010, 62, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Boczek, T.; Lisek, M.; Ferenc, B.; Kowalski, A.; Stepinski, D.; Wiktorska, M.; Zylinska, L. Plasma membrane Ca2+-ATPase isoforms composition regulates cellular pH homeostasis in differentiating PC12 cells in a manner dependent on cytosolic Ca2+ elevations. PLoS ONE 2014, 9, e102352. [Google Scholar] [CrossRef] [PubMed]
- Eaddy, A.C.; Schnellmann, R.G. Visualization and quantification of endoplasmic reticulum Ca2+ in renal cells using confocal microscopy and Fluo5F. Biochem. Biophys. Res. Commun. 2011, 404, 424–427. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.J.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Zorov, S.D.; Babenko, V.A.; Zorov, D.B. Functional Significance of the Mitochondrial Membrane Potential. Biochem. Suppl. Ser. A Membr. Cell Biol. 2018, 12, 20–26. [Google Scholar] [CrossRef]
- Zamaraeva, M.V.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells die with increased cytosolic ATP during apoptosis: A bioluminescence study with intracellular luciferase. Cell Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Bailey, K.M.; Wojtkowiak, J.W.; Ben-Nun, Y.; Bogyo, M.; Weber, E.; Moin, K.; Blum, G.; Mattingly, R.R.; Gillies, R.J.; et al. Acid-mediated tumor proteolysis: Contribution of cysteine cathepsins. Neoplasia 2013, 15, 1125–1137. [Google Scholar] [CrossRef]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Milanick, M.A. Proton fluxes associated with the Ca pump in human red blood cells. Am. J. Physiol. 1990, 258, C552–C562. [Google Scholar] [CrossRef]
- DeSantiago, J.; Batlle, D.; Khilnani, M.; Dedhia, S.; Kulczyk, J.; Duque, R.; Ruiz, J.; Pena-Rasgado, C.; Rasgado-Flores, H. Ca2+/H+ exchange via the plasma membrane Ca2+ ATPase in skeletal muscle. Front. Biosci. 2007, 12, 4641–4660. [Google Scholar] [CrossRef] [PubMed]
- James, A.D.; Richardson, D.A.; Oh, I.; Sritangos, P.; Attard, T.; Barrett, L.; Bruce, J.I.E. Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: Role of pyruvate kinase-M2 (PKM2). Br. J. Cancer 2019, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Brimacombe, K.R.; Mott, B.T.; Urban, D.J.; Hu, X.; Yang, S.-M.; Lee, T.D.; Cheff, D.M.; Kouznetsova, J.; Benavides, G.A.; et al. Discovery and Optimization of Potent, Cell-Active Pyrazole-Based Inhibitors of Lactate Dehydrogenase (LDH). J. Med. Chem. 2017, 60, 9184–9204. [Google Scholar] [CrossRef] [PubMed]
- Serganova, I.; Cohen, I.J.; Vemuri, K.; Shindo, M.; Maeda, M.; Mane, M.; Moroz, E.; Khanin, R.; Satagopan, J.; Koutcher, J.A.; et al. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PLoS ONE 2018, 13, e0203965. [Google Scholar] [CrossRef]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150. [Google Scholar] [CrossRef]
- Young, L.; Sung, J.; Masters, J.R. Detection of mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer ID | Catalogue # | Sequence |
|---|---|---|
| ATP2B1_Forward | H_ATP2B1_1 | ATCCTCTTGTCTGTAGTGTG |
| ATP2B1_Reverse | H_ATP2B1_1 | TCACCATATTTCACTTGAGC |
| ATP2B2_Forward | H_ATP2B2_1 | GATAGTGATCGTGCAGTTTG |
| ATP2B2_Reverse | H_ATP2B2_1 | AATGAATATGCACCACATCC |
| ATP2B3_Forward | H_ATP2B3_1 | CACCCACTACAAAGAGATTC |
| ATP2B3_Reverse | H_ATP2B3_1 | GTAGTATTTTGGTGGTATAGGC |
| ATP2B4_Forward | H_ATP2B4_1 | AACTCTCAGACTGGAATCATC |
| ATP2B4_Reverse | H_ATP2B4_1 | ACCTTTCTTCTTTTTCTCCC |
| PMCA4b_Forward | N/A | CCAGACTCAGATCAAAGTGGTCA |
| PMCA4b_Reverse | N/A | TCGTGGCAACTCCTCCTCTA |
| S18rRNA_Forward | H_RN18S1_1 | ATCGGGGATTGCAATTATTC |
| S18rRNA _Reverse | H_RN18S1_1 | CTCACTAAACCATCCAATCG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sritangos, P.; Pena Alarcon, E.; James, A.D.; Sultan, A.; Richardson, D.A.; Bruce, J.I.E. Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers 2020, 12, 218. https://doi.org/10.3390/cancers12010218
Sritangos P, Pena Alarcon E, James AD, Sultan A, Richardson DA, Bruce JIE. Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers. 2020; 12(1):218. https://doi.org/10.3390/cancers12010218
Chicago/Turabian StyleSritangos, Pishyaporn, Eduardo Pena Alarcon, Andrew D. James, Ahlam Sultan, Daniel A. Richardson, and Jason I. E. Bruce. 2020. "Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer" Cancers 12, no. 1: 218. https://doi.org/10.3390/cancers12010218
APA StyleSritangos, P., Pena Alarcon, E., James, A. D., Sultan, A., Richardson, D. A., & Bruce, J. I. E. (2020). Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers, 12(1), 218. https://doi.org/10.3390/cancers12010218