Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy


Abstract
1. Introduction to Heat Shock Proteins/Chaperones
2. HSP Chaperones and Cancer
3. HSP90
3.1. HSP90 Structure and Functions
3.2. HSP90 and Nonfusion Protein Kinases
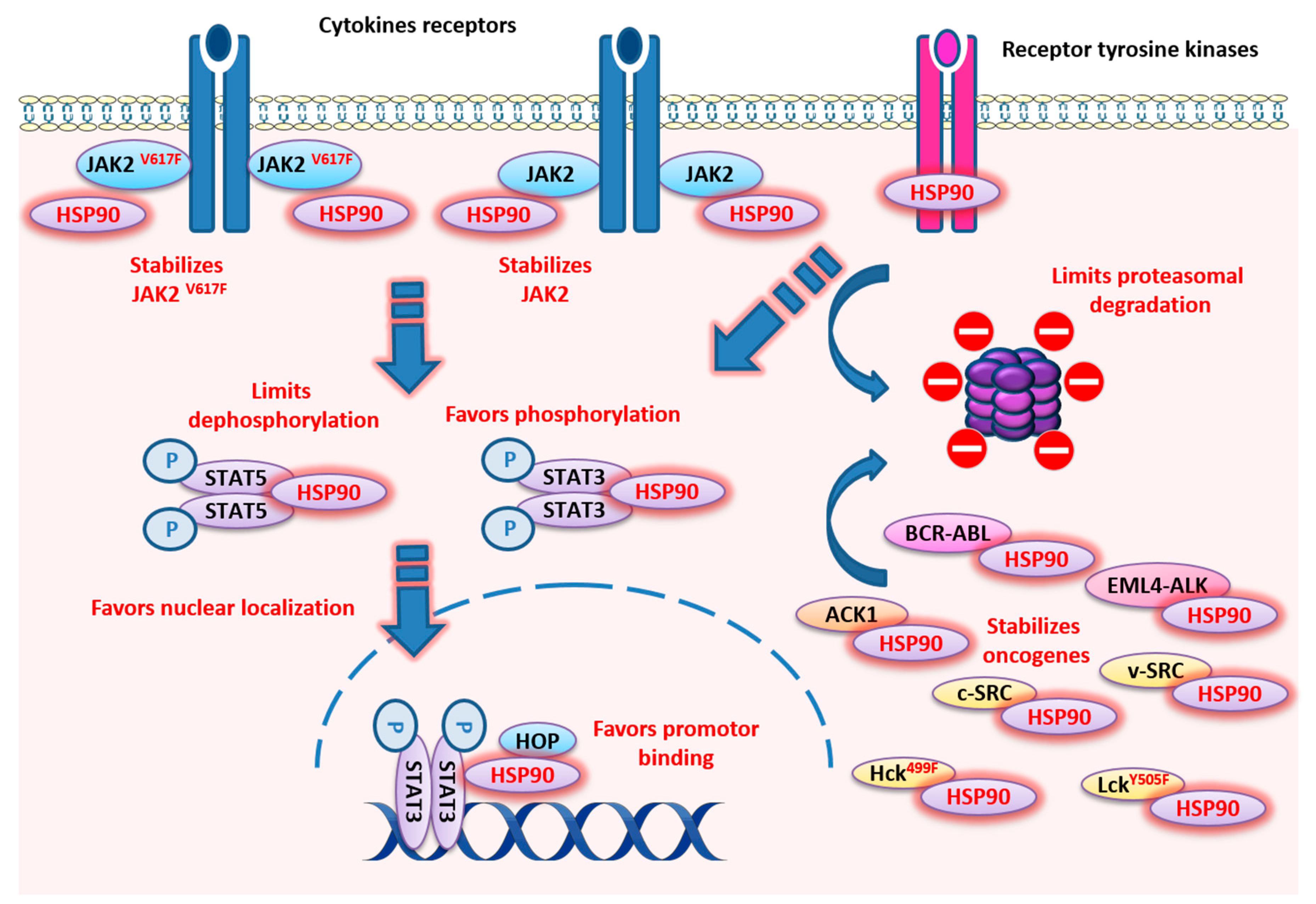
3.2.1. Jak Kinases
3.2.2. Src Kinases
3.2.3. ACK1
3.2.4. BRAF
3.3. HSP90 and Fusion Protein Kinases
3.3.1. BCR-ABL
3.3.2. EML4-ALK
3.4. HSP90 and ErbB Family of Receptor Tyrosine Kinase (RTK)
3.5. STAT3/5 and HSP90
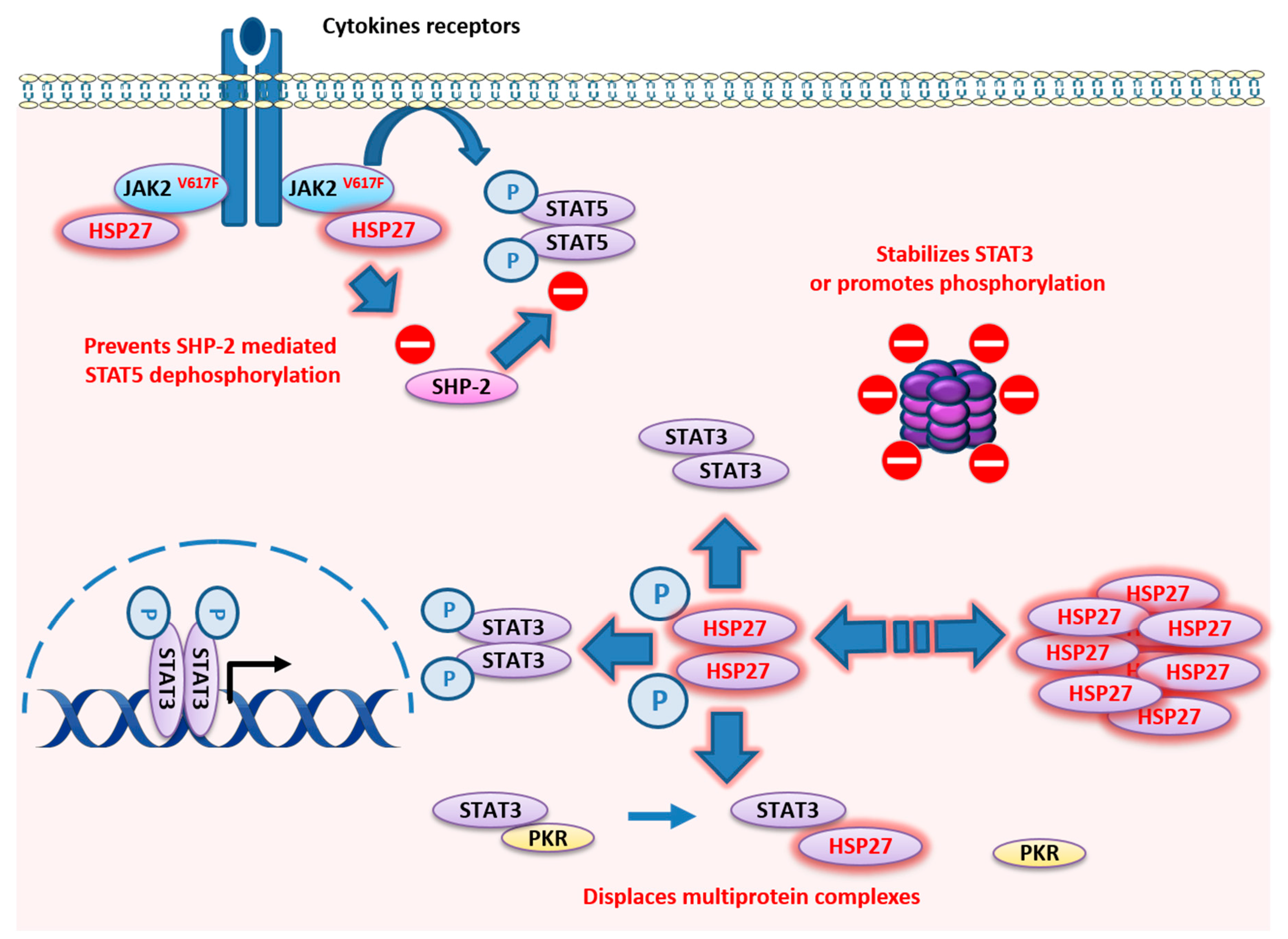
4. HSP27
4.1. HSP27 Structure
4.2. HSP27 Functions
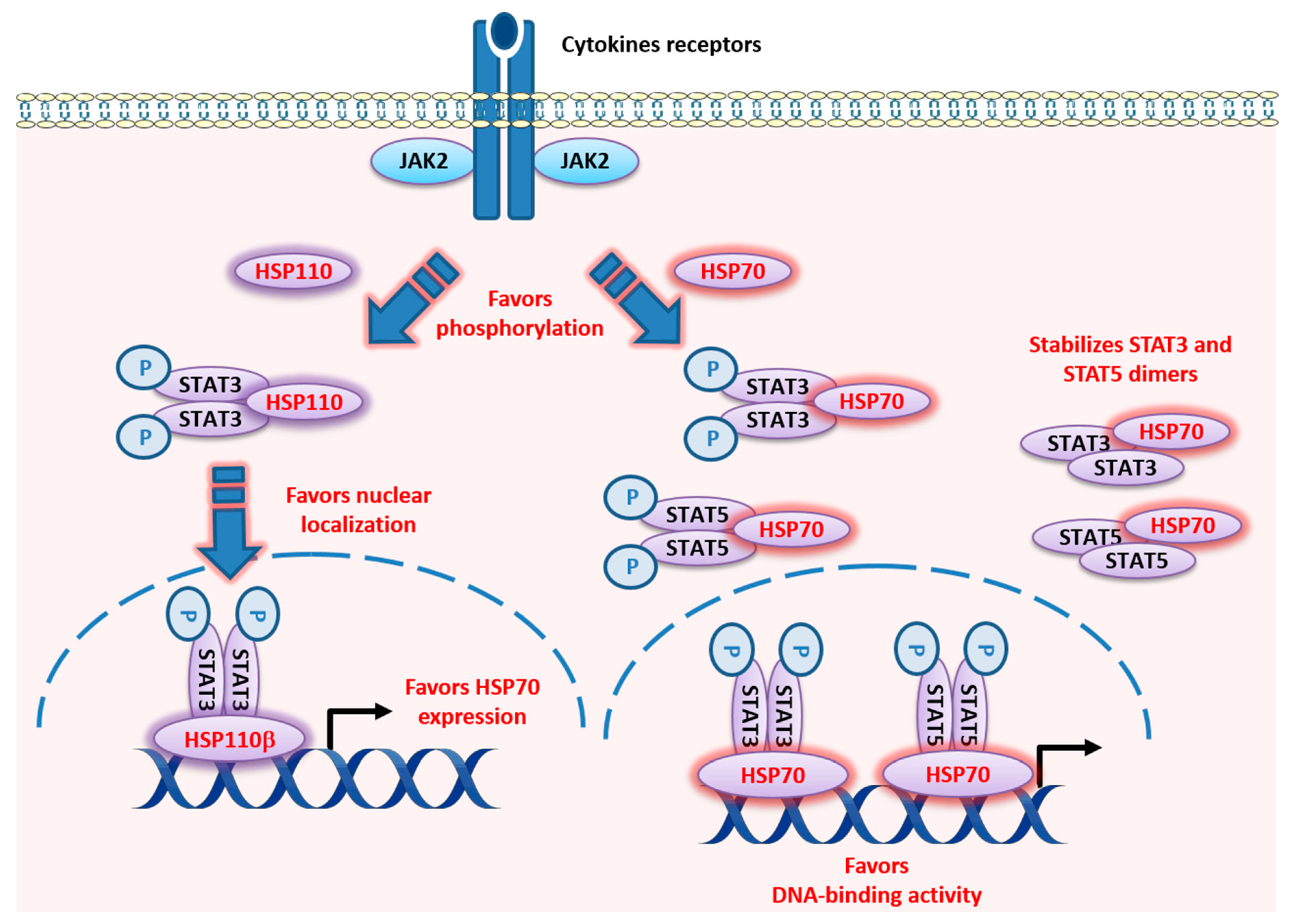
5. HSP110
5.1. HSP110 Structure
5.2. HSP110 Functions
6. HSP70
6.1. HSP70 Structure
6.2. HSP70 Functions
7. Regulation Mechanisms of HSF/HSPs by JAK/STAT Signaling: A Feedback Loop?
7.1. HSF and SOCS Regulation
7.2. HSF/HSPs and JAK/STAT
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sorger, P.K. Heat shock factor and the heat shock response. Cell 1991, 65, 363–366. [Google Scholar] [CrossRef]
- Welch, W.J. Heat shock proteins functioning as molecular chaperones: Their roles in normal and stressed cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1993, 339, 327–333. [Google Scholar] [PubMed]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends BioChem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Lianos, G.D.; Alexiou, G.A.; Mangano, A.; Mangano, A.; Rausei, S.; Boni, L.; Dionigi, G.; Roukos, D.H. The role of heat shock proteins in cancer. Cancer Lett. 2015, 360, 114–118. [Google Scholar] [CrossRef]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Guttmann, D.M.; Koumenis, C. The heat shock proteins as targets for radiosensitization and chemosensitization in cancer. Cancer Biol. Ther. 2011, 12, 1023–1031. [Google Scholar] [CrossRef]
- Chi, K.N.; Yu, E.Y.; Jacobs, C.; Bazov, J.; Kollmannsberger, C.; Higano, C.S.; Mukherjee, S.D.; Gleave, M.E.; Stewart, P.S.; Hotte, S.J. A phase I dose-escalation study of apatorsen (OGX-427), an antisense inhibitor targeting heat shock protein 27 (Hsp27), in patients with castration-resistant prostate cancer and oTher. advanced cancers. Ann. Oncol. 2016, 27, 1116–1122. [Google Scholar] [CrossRef]
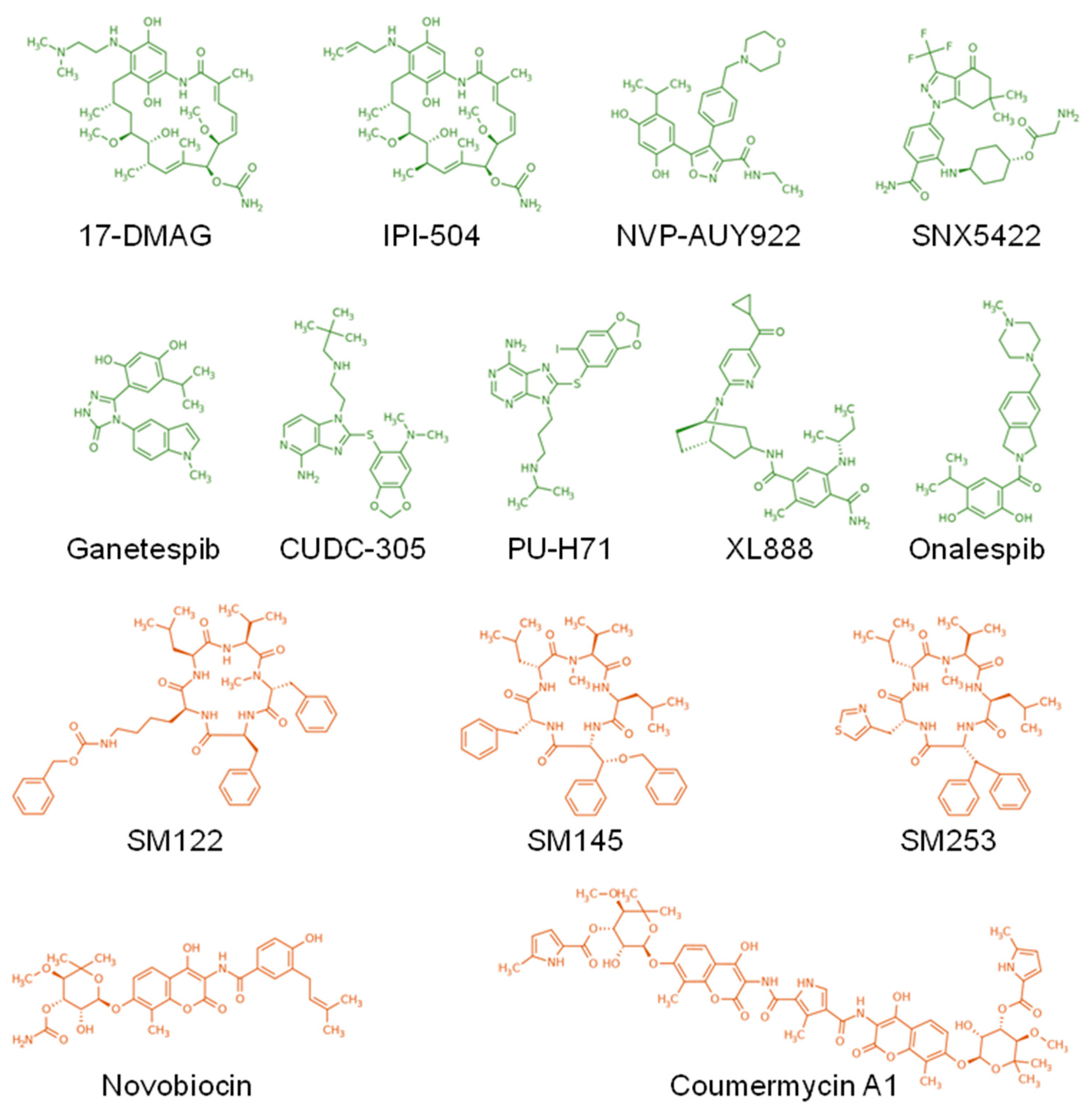
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Yuno, A.; Lee, M.J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Methods Mol. Biol. 2018, 1709, 423–441. [Google Scholar] [PubMed]
- Rocchi, P.; So, A.; Kojima, S.; Signaevsky, M.; Beraldi, E.; Fazli, L.; Hurtado-Coll, A.; Yamanaka, K.; Gleave, M. Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer Res. 2004, 64, 6595–6602. [Google Scholar] [CrossRef] [PubMed]
- Song, T.F.; Zhang, Z.F.; Liu, L.; Yang, T.; Jiang, J.; Li, P. Small interfering RNA-mediated silencing of heat shock protein 27 (HSP27) Increases chemosensitivity to paclitaxel by increasing production of reactive oxygen species in human ovarian cancer cells (HO8910). J. Int. Med. Res. 2009, 37, 1375–1388. [Google Scholar] [CrossRef]
- Baylot, V.; Andrieu, C.; Katsogiannou, M.; Taieb, D.; Garcia, S.; Giusianom, S.; Acunzo, J.; Iovanna, J.; Gleave, M.; Garrido, C.; et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011, 2, e221. [Google Scholar] [CrossRef]
- Lelj-Garolla, B.; Kumano, M.; Beraldi, E.; Nappi, L.; Rocchi, P.; Ionescu, D.N.; Fazlim, L.; Zoubeidi, A.; Gleave, M.E. Hsp27 Inhibition with OGX-427 Sensitizes Non-Small Cell Lung Cancer Cells to Erlotinib and Chemotherapy. Mol. Cancer Ther. 2015, 14, 1107–1116. [Google Scholar] [CrossRef]
- Spigel, D.R.; Shipley, D.L.; Waterhouse, D.M.; Jones, S.F.; Ward, P.J.; Shih, K.C.; Hemphill, B.; McCleod, M.; Whorf, R.C.; Page, R.D.; et al. A Randomized Double-Blinded Phase II Trial of Carboplatin and Pemetrexed with or without Apatorsen (OGX-427) in Patients with Previously Untreated Stage IV Non-Squamous-Non-Small-Cell Lung Cancer: The SPRUCE Trial. Oncologist 2019, 24, e1409–e1416. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, Y.; Wan, J.; Wang, M.; Rocchi, P.; Qu, F.; Iovanna, J.L.; Peng, L. Novel triazole ribonucleoside down-regulates heat shock protein 27 and induces potent anticancer activity on drug-resistant pancreatic cancer. J. Med. Chem. 2009, 52, 6083–6096. [Google Scholar] [CrossRef]
- Chauhan, D.; Li, G.; Shringarpure, R.; Podar, K.; Ohtake, Y.; Hideshima, T.; Anderson, K.C. Blockade of Hsp27 overcomes Bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res. 2003, 63, 6174–6177. [Google Scholar]
- Heinrich, J.C.; Tuukkanen, A.; Schroeder, M.; Fahrig, T.; Fahrig, R. RP101 (brivudine) binds to heat shock protein HSP27 (HSPB1) and enhances survival in animals and pancreatic cancer patients. J. Cancer Res. Clin. Oncol. 2011, 137, 1349–1361. [Google Scholar] [CrossRef]
- Kaiser, M.; Kühnl, A.; Reins, J.; Fischer, S.; Ortiz-Tanchez, J.; Schlee, C.; Mochmann, L.H.; Heesch, S.; Benlasfer, O.; Hofmann, W.K.; et al. Antileukemic activity of the HSP70 inhibitor pifithrin-μ in acute leukemia. Blood Cancer J. 2011, 1, e28. [Google Scholar] [CrossRef] [PubMed]
- Goloudina, A.R.; Demidov, O.N.; Garrido, C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012, 325, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer ChemoTher. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Brünnert, D.; Langer, C.; Zimmermann, L.; Bargou, R.C.; Burchardt, M.; Chatterjee, M.; Stope, M.B. The heat shock protein 70 inhibitor VER155008 suppresses the expression of HSP27, HOP and HSP90β and the androgen receptor, induces apoptosis, and attenuates prostate cancer cell growth. J. Cell BioChem. 2020, 121, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Andrulis, M.; Stühmer, T.; Müller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Rérole, A.L.; Gobbo, J.; De Thonel, A.; Schmitt, E.; Pais de Barros, J.P.; Hammann, A.; Lanneau, D.; Fourmaux, E.; Demidov, O.N.; Micheau, O.; et al. Peptides and aptamers targeting HSP70: A novel approach for anticancer chemotherapy. Cancer Res. 2011, 71, 484–495. [Google Scholar] [CrossRef]
- Schmitt, E.; Maingret, L.; Puig, P.E.; Rerole, A.L.; Ghiringhelli, F.; Hammann, A.; Solary, E.; Kroemer, G.; Garrido, C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006, 66, 4191–4197. [Google Scholar] [CrossRef]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef]
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: A clinical phase i trial. Clin. Cancer Res. 2004, 10, 3699–3707. [Google Scholar] [CrossRef]
- Shiotsu, Y.; Neckers, L.M.; Wortman, I.; An, W.G.; Schulte, T.W.; Soga, S.; Murakata, C.; Tamaoki, T.; Akinaga, S. Novel oxime derivatives of radicicol induce erythroid differentiation associated with preferential G (1) phase accumulation against chronic myelogenous leukemia cells through destabilization of Bcr-Abl with Hsp90 complex. Blood 2000, 96, 2284–2291. [Google Scholar] [CrossRef]
- Palacios, C.; López-Pérez, A.I.; López-Rivas, A. Down-regulation of RIP expression by 17-dimethylaminoethylamino-17-demethoxygeldanamycin promotes TRAIL-induced apoptosis in breast tumor cells. Cancer Lett. 2010, 287, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S.; Svenstrup, T.H.; Guerra, B. The small-molecule kinase inhibitor D11 counteracts 17-AAG-mediated up-regulation of HSP70 in brain cancer cells. PLoS ONE 2017, 12, e0177706. [Google Scholar] [CrossRef] [PubMed]
- Ayrault, O.; Godeny, M.D.; Dillon, C.; Zindy, F.; Fitzgerald, P.; Roussel, M.F.; Beere, H.M. Inhibition of Hsp90 via 17-DMAG induces apoptosis in a p53-dependent manner to prevent medulloblastoma. Proc. Natl. Acad. Sci. USA 2009, 106, 17037–17042. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.L.; Gupta, N.; Lehman, A.; Ruppert, A.S.; Yu, L.; Oakes, C.C.; Claus, R.; Plass, C.; Maddocks, K.J.; Andritsos, L.; et al. Hsp90 inhibition increases SOCS3 transcript and regulates migration and cell death in chronic lymphocytic leukemia. Oncotarget 2016, 7, 28684–28696. [Google Scholar] [CrossRef]
- Hanson, B.E.; Vesole, D.H. Retaspimycin hydrochloride (IPI-504): A novel heat shock protein inhibitor as an anticancer agent. Expert Opin. Investig. Drugs 2009, 18, 1375–1383. [Google Scholar] [CrossRef]
- Song, D.; Chaerkady, R.; Tan, A.C.; García-García, E.; Nalli, A.; Suárez-Gauthier, A.; López-Ríos, F.; Zhang, X.F.; Solomon, A.; Tong, J.; et al. Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 3275–3284. [Google Scholar] [CrossRef]
- Wagner, A.J.; Chugh, R.; Rosen, L.S.; Morgan, J.A.; George, S.; Gordon, M.; Dunbar, J.; Normant, E.; Grayzel, D.; Demetri, G.D.; et al. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin. Cancer Res. 2013, 19, 6020–6029. [Google Scholar] [CrossRef]
- Scaltriti, M.; Serra, V.; Normant, E.; Guzman, M.; Rodriguez, O.; Lim, A.R.; Slocum, K.L.; West, K.A.; Rodriguez, V.; Prudkin, L.; et al. Antitumor activity of the Hsp90 inhibitor IPI-504 in HER2-positive trastuzumab-resistant breast cancer. Mol. Cancer Ther. 2011, 10, 817–824. [Google Scholar] [CrossRef]
- Floris, G.; Debiec-Rychter, M.; Wozniak, A.; Stefan, C.; Normant, E.; Faa, G.; Machiels, K.; Vanleeuw, U.; Sciot, R.; Schöffski, P. The heat shock protein 90 inhibitor IPI-504 induces KIT degradation, tumor shrinkage, and cell proliferation arrest in xenograft models of gastrointestinal stromal tumors. Mol. Cancer Ther. 2011, 10, 1897–1908. [Google Scholar] [CrossRef]
- Normant, E.; Paez, G.; West, K.A.; Lim, A.R.; Slocum, K.L.; Tunkey, C.; McDougall, J.; Wylie, A.A.; Robison, K.; Caliri, K.; et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 2011, 30, 2581–2586. [Google Scholar] [CrossRef]
- Hendriks, L.E.L.; Dingemans, A.C. Heat shock protein antagonists in early stage clinical trials for NSCLC. Expert Opin. Investig. Drugs 2017, 26, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.X.; Xu, J.H.; Zhang, K.Z.; Lin, Q.; Huang, X.W.; Wen, C.X.; Chen, Y.Z. Disruption of the Bcr-Abl/Hsp90 protein complex: A possible mechanism to inhibit Bcr-Abl-positive human leukemic blasts by novobiocin. Leukemia 2008, 22, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Shelton, S.N.; Shawgo, M.E.; Matthews, S.B.; Lu, Y.; Donnelly, A.C.; Szabla, K.; Tanol, M.; Vielhauer, G.A.; Rajewski, R.A.; Matts, R.L.; et al. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol. 2009, 76, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.B.; Vielhauer, G.A.; Manthe, C.A.; Chaguturu, V.K.; Szabla, K.; Matts, R.L.; Donnelly, A.C.; Blagg, B.S.; Holzbeierlein, J.M. Characterization of a novel novobiocin analogue as a putative C-terminal inhibitor of heat shock protein 90 in prostate cancer cells. Prostate 2010, 70, 27–36. [Google Scholar] [CrossRef]
- Le, H.T.; Vielhauer, G.A.; Manthe, C.A.; Chaguturu, V.K.; Szabla, K.; Matts, R.L.; Donnelly, A.C.; Blagg, B.S.; Holzbeierlein, J.M. Panaxynol, a natural Hsp90 inhibitor, effectively targets both lung cancer stem and non-stem cells. Cancer Lett. 2018, 412, 297–307. [Google Scholar] [CrossRef]
- Lin, S.F.; Lin, J.D.; Hsueh, C.; Chou, T.C.; Yeh, C.N.; Chen, M.H.; Wong, R.J. Efficacy of an HSP90 inhibitor, ganetespib, in preclinical thyroid cancer models. Oncotarget 2017, 8, 41294–41304. [Google Scholar] [CrossRef]
- Lee, H.; Saini, N.; Howard, E.W.; Parris, A.B.; Ma, Z.; Zhao, Q.; Zhao, M.; Liu, B.; Edgerton, S.M.; Thor, A.D.; et al. Ganetespib targets multiple levels of the receptor tyrosine kinase signaling cascade and preferentially inhibits ErbB2-overexpressing breast cancer cells. Sci. Rep. 2018, 8, 6829. [Google Scholar] [CrossRef]
- He, W.; Hu, H. BIIB021, an Hsp90 inhibitor: A promising therapeutic strategy for blood malignancies (Review). Oncol. Rep. 2018, 40, 3–15. [Google Scholar] [CrossRef]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gönen, M.; Gozman, A.; et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Investig. 2010, 120, 3578–3593. [Google Scholar] [CrossRef]
- Fiskus, W.; Verstovsek, S.; Manshouri, T.; Rao, R.; Balusu, R.; Venkannagari, S.; Rao, N.N.; Ha, K.; Smith, J.E.; Hembruff, S.L.; et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin. Cancer Res. 2011, 17, 7347–7358. [Google Scholar] [CrossRef]
- Park, K.S.; Hong, Y.S.; Choi, J.; Yoon, S.; Kang, J.; Kim, D.; Lee, K.P.; Im, H.S.; Lee, C.H.; Seo, S.; et al. HSP90 inhibitor, AUY922, debilitates intrinsic and acquired lapatinib-resistant HER2-positive gastric cancer cells. BMB Rep. 2018, 51, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, M.H.; Park, I.; Jeon, H.; Choi, J.; Seo, S.; Kim, S.W.; Koh, G.Y.; Park, K.S.; Lee, D.H. HSP90 inhibitor (NVP-AUY922) enhances the anti-cancer effect of BCL-2 inhibitor (ABT-737) in small cell lung cancer expressing BCL-2. Cancer Lett. 2017, 411, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. The dipeptidyl peptidase-IV inhibitor gemigliptin alone or in combination with NVP-AUY922 has a cytotoxic activity in thyroid carcinoma cells. Tumour. Biol. 2017, 39, 1010428317722068. [Google Scholar] [CrossRef] [PubMed]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.M.; Collins, I.; Davies, N.G.; Drysdale, M.J.; et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, W.; Dong, W.; Wang, Z.; Qin, Y.; Zhang, T.; Zhang, H. HSP90 inhibitor NVP-AUY922 induces cell apoptosis by disruption of the survivin in papillary thyroid carcinoma cells. Biochem. Biophys. Res. Commun. 2017, 487, 313–319. [Google Scholar] [CrossRef]
- Tao, W.; Chakraborty, S.N.; Leng, X.; Ma, H.; Arlinghaus, R.B. HSP90 inhibitor AUY922 induces cell death by disruption of the Bcr-Abl, Jak2 and HSP90 signaling network complex in leukemia cells. Genes Cancer 2015, 6, 19–29. [Google Scholar]
- Chakraborty, S.N.; Leng, X.; Perazzona, B.; Sun, X.; Lin, Y.H.; Arlinghaus, R.B. Combination of JAK2 and HSP90 inhibitors: An effective therapeutic option in drug-resistant chronic myelogenous leukemia. Genes Cancer 2016, 7, 201–208. [Google Scholar]
- Hobbs, G.S.; Hanasoge Somasundara, A.V.; Kleppe, M.; Litvin, R.; Arcila, M.; Ahn, J.; McKenney, A.S.; Knapp, K.; Ptashkin, R.; Weinstein, H.; et al. Hsp90 inhibition disrupts JAK-STAT signaling and leads to reductions in splenomegaly in patients with myeloproliferative neoplasms. Haematologica 2018, 103, e5–e9. [Google Scholar] [CrossRef]
- Johnson, M.L.; Yu, H.A.; Hart, E.M.; Weitner, B.B.; Rademaker, A.W.; Patel, J.D.; Kris, M.G.; Riely, G.J. Phase I/II Study of HSP90 Inhibitor AUY922 and Erlotinib for EGFR-Mutant Lung Cancer with Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J. Clin. Oncol. 2015, 33, 1666–1673. [Google Scholar] [CrossRef]
- Weigert, O.; Lane, A.A.; Bird, L.; Kopp, N.; Chapuy, B.; van Bodegom, D.; Toms, A.V.; Marubayashi, S.; Christie, A.L.; McKeown, M.; et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J. Exp. Med. 2012, 209, 259–273. [Google Scholar] [CrossRef]
- Mahendrarajah, N.; Borisova, M.E.; Reichardt, S.; Godmann, M.; Sellmer, A.; Mahboobi, S.; Haitel, A.; Schmid, K.; Kenner, L.; Heinzel, T.; et al. HSP90 is necessary for the ACK1-dependent phosphorylation of STAT1 and STAT3. Cell Signal. 2017, 39, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Paraiso, K.H.T.; Hearn, K.; Rodriguez-Lopez, A.M.; Munck, J.M.; Haarberg, H.E.; Sondak, V.K.; Thompson, N.T.; Azab, M.; Lyons, J.F.; et al. Inhibition of HSP90 by AT13387 delays the emergence of resistance to BRAF inhibitors and overcomes resistance to dual BRAF and MEK inhibition in melanoma models. Mol. Cancer Ther. 2014, 13, 2793–2804. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.; Curry, J.; Smyth, T.; Fazal, L.; Feltell, R.; Harada, I.; Coyle, J.; Williams, B.; Reule, M.; Angove, H.; et al. The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012, 103, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Courtin, A.; Smyth, T.; Hearn, K.; Saini, H.K.; Thompson, N.T.; Lyons, J.F.; Wallis, N.G. Emergence of resistance to tyrosine kinase inhibitors in non-small-cell lung cancer can be delayed by an upFront combination with the HSP90 inhibitor onalespib. Br. J. Cancer 2016, 115, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Chen, Y.A.; Gibney, G.T.; Weber, J.S.; Kudchadkar, R.R.; Khushalani, N.I.; Markowitz, J.; Brohl, A.S.; Tetteh, L.F.; Ramadan, H.; et al. Combined BRAF and HSP90 Inhibition in Patients with Unresectable. Clin. Cancer Res. 2018, 24, 5516–5524. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.A.; Wise, S.C.; Hu, M.; Gouveia, C.; Vander Broek, R.; Freudlsperger, C.; Kannabiran, V.R.; Arun, P.; Mitchell, J.B.; Chen, Z.; et al. HSP90 Inhibitor SNX5422/2112 Targets the Dysregulated Signal and Transcription Factor Network and Malignant Phenotype of Head and Neck Squamous Cell Carcinoma. Transl. Oncol. 2013, 6, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.W.; Veal, J.M.; Barabasz, A.; Foley, B.; Fadden, P.; Scott, A.; Huang, K.; Steed, P.; Hall, S. Targeting of multiple signaling pathways by the Hsp90 inhibitor SNX-2112 in EGFR resistance models as a single agent or in combination with erlotinib. Oncol. Res. 2009, 18, 229–242. [Google Scholar] [CrossRef]
- Smith, D.L.; Acquaviva, J.; Sequeira, M.; Jimenez, J.P.; Zhang, C.; Sang, J.; Bates, R.C.; Proia, D.A. The HSP90 inhibitor ganetespib potentiates the antitumor activity of EGFR tyrosine kinase inhibition in mutant and wild-type non-small cell lung cancer. Target. Oncol. 2015, 10, 235–245. [Google Scholar] [CrossRef]
- Moulick, K.; Ahn, J.H.; Zong, H.; Rodina, A.; Cerchietti, L.; Gomes DaGama, E.M.; Caldas-Lopes, E.; Beebe, K.; Perna, F.; Hatzi, K.; et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 2011, 7, 818–826. [Google Scholar] [CrossRef]
- Hong, Y.S.; Jang, W.J.; Chun, K.S.; Jeong, C.H. Hsp90 inhibition by WK88-1 potently suppresses the growth of gefitinib-resistant H1975 cells harboring the T790M mutation in EGFR. Oncol. Rep. 2014, 31, 2619–2624. [Google Scholar] [CrossRef][Green Version]
- Kobayashi, N.; Toyooka, S.; Soh, J.; Yamamoto, H.; Dote, H.; Kawasaki, K.; Otani, H.; Kubo, T.; Jida, M.; Ueno, T.; et al. The anti-proliferative effect of heat shock protein 90 inhibitor, 17-DMAG, on non-small-cell lung cancers being resistant to EGFR tyrosine kinase inhibitor. Lung Cancer 2012, 75, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, G.J.; Gonzalez, D.; Boudesco, C.; Dias, A.M.M.; Gotthard, G.; Uyanik, B.; Dondaine, L.; Marcion, G.; Hermetet, F.; Denis, C.; et al. Selecting the first chemical molecule inhibitor of HSP110 for colorectal cancer therapy. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Xia, Y.; Deutschbauer, A.M.; Davis, R.W.; Gerstein, M.; Frydman, J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 2007, 131, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Makhnevych, T.; Houry, W.A. The role of Hsp90 in protein complex assembly. Biochim. Biophys. Acta 2012, 1823, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Boczek, E.E.; Reefschläger, L.G.; Dehling, M.; Struller, T.J.; Häusler, E.; Seidl, A.; Kaila, V.R.; Buchner, J. Conformational processing of oncogenic v-Src kinase by the molecular chaperone Hsp90. Proc. Natl. Acad. Sci. USA 2015, 112, E3189–E3198. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.J.; Prince, T.; Chengm, J.; Stevenson, M.A.; Calderwood, S.K. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 2008, 8, 491–495. [Google Scholar] [CrossRef]
- Shang, L.; Tomasi, T.B. The heat shock protein 90-CDC37 chaperone complex is required for signaling by types I and II interferons. J. Biol. Chem. 2006, 281, 1876–1884. [Google Scholar] [CrossRef]
- Peeters, P.; Wlodarska, I.; Baens, M.; Criel, A.; Selleslag, D.; Hagemeijer, A.; Van den Berghe, H.; Marynen, P. Fusion of ETV6 to MDS1/EVI1 as a result of t(3;12)(q26;p13) in myeloproliferative disorders. Cancer Res. 1997, 57, 564–569. [Google Scholar]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffé, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Levine, R.L.; Loriaux, M.; Huntly, B.J.; Loh, M.L.; Beran, M.; Stoffregen, E.; Berger, R.; Clark, J.J.; Willis, S.G.; Nguyen, K.T.; et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid Leukemia but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 2005, 106, 3377–3379. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Kreil, S.; Zoi, K.; Waghorn, K.; Curtis, C.; Zhang, L.; Score, J.; Seear, R.; Chase, A.J.; Grand, F.H.; et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005, 106, 2162–2168. [Google Scholar] [CrossRef]
- Bareng, J.; Jilani, I.; Gorre, M.; Kantarjian, H.; Giles, F.; Hannah, A.; Albitar, M. A potential role for HSP90 inhibitors in the treatment of JAK2 mutant-positive diseases as demonstrated using quantitative flow cytometry. Leuk. Lymphoma 2007, 48, 2189–2195. [Google Scholar] [CrossRef]
- Sen, B.; Johnson, F.M. Regulation of SRC family kinases in human cancers. J. Signal. Transduct 2011, 2011, 865819. [Google Scholar] [CrossRef]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef]
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Takeya, T.; Hanafusa, H. Structure and sequence of the cellular gene homologous to the RSV src gene and the mechanism for generating the transforming virus. Cell 1983, 32, 881–890. [Google Scholar] [CrossRef]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef]
- Hunter, T.; Sefton, B.M. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. USA 1980, 77, 1311–1315. [Google Scholar] [CrossRef]
- Xu, Y.; Lindquist, S. Heat-shock protein hsp90 governs the activity of pp60v-src kinase. Proc. Natl. Acad. Sci. USA 1993, 90, 7074–7078. [Google Scholar] [CrossRef]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef]
- Nathan, D.F.; Lindquist, S. Mutational analysis of Hsp90 function: Interactions with a steroid receptor and a protein kinase. Mol. Cell Biol. 1995, 15, 3917–3925. [Google Scholar] [CrossRef]
- Luo, Q.; Boczek, E.E.; Wang, Q.; Buchner, J.; Kaila, V.R. Hsp90 dependence of a kinase is determined by its conformational landscape. Sci. Rep. 2017, 7, 43996. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef]
- Giannini, A.; Bijlmakers, M.J. Regulation of the Src family kinase Lck by Hsp90 and ubiquitination. Mol. Cell Biol. 2004, 24, 5667–5676. [Google Scholar] [CrossRef]
- Scholz, G.M.; Hartson, S.D.; Cartledge, K.; Volk, L.; Matts, R.L.; Dunn, A.R. The molecular chaperone Hsp90 is required for signal transduction by wild-type Hck and maintenance of its constitutively active counterpart. Cell Growth Differ. 2001, 12, 409–417. [Google Scholar]
- Mahajan, N.P.; Whang, Y.E.; Mohler, J.L.; Earp, H.S. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: Role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005, 65, 10514–10523. [Google Scholar] [CrossRef]
- Karachaliou, N.; Pilotto, S.; Teixidó, C.; Viteri, S.; González-Cao, M.; Riso, A.; Morales-Espinosa, D.; Molina, M.A.; Chaib, I.; Santarpia, M.; et al. Melanoma: Oncogenic drivers and the immune system. Ann. Transl. Med. 2015, 3, 265. [Google Scholar]
- Da Rocha Dias, S.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef]
- Coppo, P.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 2006, 134, 171–179. [Google Scholar] [CrossRef]
- Nair, R.R.; Tolentino, J.H.; Hazlehurst, L.A. Role of STAT3 in Transformation and Drug Resistance in CML. Front. Oncol. 2012, 2, 30. [Google Scholar] [CrossRef]
- De Groot, R.P.; Raaijmakers, J.A.; Lammers, J.W.; Jove, R.; Koenderman, L. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood 1999, 94, 1108–1112. [Google Scholar] [CrossRef]
- Sillaber, C.; Gesbert, F.; Frank, D.A.; Sattler, M.; Griffin, J.D. STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood 2000, 95, 2118–2125. [Google Scholar] [CrossRef]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef]
- Esfahani, K.; Cohen, V. HSP90 as a novel molecular target in non-small-cell lung cancer. Lung Cancer (Auckl) 2016, 7, 11–17. [Google Scholar]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Kosack, L.; Wingelhofer, B.; Popa, A.; Orlova, A.; Agerer, B.; Vilagos, B.; Majek, P.; Parapatics, K.; Lercher, A.; Ringler, A.; et al. The ERBB-STAT3 Axis Drives Tasmanian Devil Facial Tumor Disease. Cancer Cell 2019, 35, 125–139.e9. [Google Scholar] [CrossRef]
- Xu, W.; Mimnaugh, E.G.; Kim, J.S.; Trepel, J.B.; Neckers, L.M. Hsp90, not Grp94, regulates the intracellular trafficking and stability of nascent ErbB2. Cell Stress Chaperones 2002, 7, 91–96. [Google Scholar] [CrossRef]
- Peng, X.; Guo, X.; Borkan, S.C.; Bharti, A.; Kuramochi, Y.; Calderwood, S.; Sawyer, D.B. Heat shock protein 90 stabilization of ErbB2 expression is disrupted by ATP depletion in myocytes. J. Biol. Chem. 2005, 280, 13148–13152. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) 2017, 9, 52. [Google Scholar] [CrossRef]
- Bao, R.; Lai, C.J.; Wang, D.G.; Qu, H.; Yin, L.; Zifcak, B.; Tao, X.; Wang, J.; Atoyan, R.; Samson, M.; et al. Targeting heat shock protein 90 with CUDC-305 overcomes erlotinib resistance in non-small cell lung cancer. Mol. Cancer Ther. 2009, 8, 3296–3306. [Google Scholar] [CrossRef]
- Ono, N.; Yamazaki, T.; Tsukaguchi, T.; Fujii, T.; Sakata, K.; Suda, A.; Tsukuda, T.; Mio, T.; Ishii, N.; Kondoh, O.; et al. Enhanced antitumor activity of erlotinib in combination with the Hsp90 inhibitor CH5164840 against non-small-cell lung cancer. Cancer Sci. 2013, 104, 1346–1352. [Google Scholar] [CrossRef]
- Sato, N.; Yamamoto, T.; Sekine, Y.; Yumioka, T.; Junicho, A.; Fuse, H.; Matsuda, T. Involvement of heat-shock protein 90 in the interleukin-6-mediated signaling pathway through STAT3. Biochem. Biophys. Res. Commun. 2003, 300, 847–852. [Google Scholar] [CrossRef]
- Song, S.; Su, Z.; Xu, H.; Niu, M.; Chen, X.; Min, H.; Zhang, B.; Sun, G.; Xie, S.; Wang, H.; et al. Luteolin selectively kills STAT3 highly activated gastric cancer cells through enhancing the binding of STAT3 to SHP-1. Cell Death Dis. 2017, 8, e2612. [Google Scholar] [CrossRef]
- Longshaw, V.M.; Baxter, M.; Prewitz, M.; Blatch, G.L. Knockdown of the co-chaperone Hop promotes extranuclear accumulation of Stat3 in mouse embryonic stem cells. Eur. J. Cell Biol. 2009, 88, 153–166. [Google Scholar] [CrossRef]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef]
- Echeverría, P.C.; Mazaira, G.; Erlejman, A.; Gomez-Sanchez, C.; Piwien Pilipuk, G.; Galigniana, M.D. Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol. Cell Biol. 2009, 29, 4788–4797. [Google Scholar] [CrossRef]
- Adwan, T.S.; Ohm, A.M.; Jones, D.N.; Humphries, M.J.; Reyland, M.E. Regulated binding of importin-α to protein kinase Cδ in response to apoptotic signals facilitates nuclear import. J. Biol. Chem. 2011, 286, 35716–35724. [Google Scholar] [CrossRef] [PubMed]
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. The Stat3/GR interaction code: Predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell 2012, 47, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Engblom, D.; Kornfeld, J.W.; Schwake, L.; Tronche, F.; Reimann, A.; Beug, H.; Hennighausen, L.; Moriggl, R.; Schütz, G. Direct glucocorticoid receptor-Stat5 interaction in hepatocytes controls body size and maturation-related gene expression. Genes Dev. 2007, 21, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Ehrnsperger, M.; Gräber, S.; Gaestel, M.; Buchner, J. Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 1997, 16, 221–229. [Google Scholar] [CrossRef]
- Shashidharamurthy, R.; Koteiche, H.A.; Dong, J.; McHaourab, H.S. Mechanism of chaperone function in small heat shock proteins: Dissociation of the HSP27 oligomer is required for recognition and binding of destabilized T4 lysozyme. J. Biol. Chem. 2005, 280, 5281–5289. [Google Scholar] [CrossRef]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers (Basel) 2019, 11, 1195. [Google Scholar] [CrossRef]
- Jehle, S.; van Rossum, B.; Stout, J.R.; Noguchi, S.M.; Falber, K.; Rehbein, K.; Oschkinat, H.; Klevit, R.E.; Rajagopal, P. alphaB-crystallin: A hybrid solid-state/solution-state NMR investigation reveals structural aspects of the heterogeneous oligomer. J. Mol. Biol. 2009, 385, 1481–1497. [Google Scholar] [CrossRef]
- Kostenko, S.; Moens, U. Heat shock protein 27 phosphorylation: Kinases, phosphatases, functions and pathology. Cell Mol. Life Sci. 2009, 66, 3289–3307. [Google Scholar] [CrossRef]
- Bruey, J.M.; Paul, C.; Fromentin, A.; Hilpert, S.; Arrigo, A.P.; Solary, E.; Garrido, C. Differential regulation of HSP27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 2000, 19, 4855–4863. [Google Scholar] [CrossRef]
- Mymrikov, E.V.; Daake, M.; Richter, B.; Haslbeck, M.; Buchner, J. The Chaperone Activity and Substrate Spectrum of Human Small Heat Shock Proteins. J. Biol. Chem. 2017, 292, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Ungelenk, S.; Moayed, F.; Ho, C.T.; Grousl, T.; Scharf, A.; Mashaghi, A.; Tans, S.; Mayer, M.P.; Mogk, A.; Bukau, B. Small heat shock proteins sequester misfolding proteins in near-native conformation for cellular protection and efficient refolding. Nat. Commun. 2016, 7, 13673. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, P.; Beraldi, E.; Ettinger, S.; Fazli, L.; Vessella, R.L.; Nelson, C.; Gleave, M. Increased Hsp27 after androgen ablation facilitates androgen-independent progression in prostate cancer via signal transducers and activators of transcription 3-mediated suppression of apoptosis. Cancer Res. 2005, 65, 11083–11093. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef]
- Shah, M.; Stanek, J.; Handwerger, S. Differential localization of heat shock proteins 90, 70, 60 and 27 in human decidua and placenta during pregnancy. HistoChem. J. 1998, 30, 509–518. [Google Scholar] [CrossRef]
- Matalon, S.T.; Drucker, L.; Fishman, A.; Ornoy, A.; Lishner, M. The Role of heat shock protein 27 in extravillous trophoblast differentiation. J. Cell BioChem. 2008, 103, 719–729. [Google Scholar] [CrossRef]
- Shochet, G.E.; Komemi, O.; Sadeh-Mestechkin, D.; Pomeranz, M.; Fishman, A.; Drucker, L.; Lishner, M.; Matalon, S.T. Heat shock protein-27 (HSP27) regulates STAT3 and eIF4G levels in first trimester human placenta. J. Mol. Histol. 2016, 47, 555–563. [Google Scholar] [CrossRef]
- Suman, P.; Malhotra, S.S.; Gupta, S.K. LIF-STAT signaling and trophoblast biology. JAKSTAT 2013, 2, e25155. [Google Scholar] [CrossRef][Green Version]
- Gibert, B.; Eckel, B.; Fasquelle, L.; Moulin, M.; Bouhallier, F.; Gonin, V.; Mellier, G.; Simon, S.; Kretz-Remy, C.; Arrigo, A.P.; et al. Knock down of heat shock protein 27 (HspB1) induces degradation of several putative client proteins. PLoS ONE 2012, 7, e29719. [Google Scholar] [CrossRef]
- Shen, L.; Qi, Z.; Zhu, Y.; Song, X.; Xuan, C.; Ben, P.; Lan, L.; Luo, L.; Yin, Z. Phosphorylated heat shock protein 27 promotes lipid clearance in hepatic cells through interacting with STAT3 and activating autophagy. Cell Signal. 2016, 28, 1086–1098. [Google Scholar] [CrossRef]
- Sevin, M.; Kubovcakova, L.; Pernet, N.; Causse, S.; Vitte, F.; Villeval, J.L.; Lacout, C.; Cordonnier, M.; Rodrigues-Lima, F.; Chanteloup, G.; et al. HSP27 is a partner of JAK2-STAT5 and a potential therapeutic target in myelofibrosis. Nat. Commun. 2018, 9, 1431. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Lee-Yoon, D.; Easton, D.; Murawski, M.; Burd, R.; Subjeck, J.R. Identification of a major subfamily of large hsp70-like proteins through the cloning of the mammalian 110-kDa heat shock protein. J. Biol. Chem. 1995, 270, 15725–15733. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Easton, D.; Murawski, M.; Kaneko, Y.; Subjeck, J.R. The chaperoning activity of hsp110. Identification of functional domains by use of targeted deletions. J. Biol. Chem. 1999, 274, 15712–15718. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, R.U.; Sharma, S.K.; Priya, S.; Finka, A.; Goloubinoff, P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 2013, 288, 21399–21411. [Google Scholar] [CrossRef] [PubMed]
- Rampelt, H.; Kirstein-Miles, J.; Nillegoda, N.B.; Chi, K.; Scholz, S.R.; Morimoto, R.I.; Bukau, B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012, 31, 4221–4235. [Google Scholar] [CrossRef]
- Wang, L.; Duke, L.; Zhang, P.S.; Arlinghaus, R.B.; Symmans, W.F.; Sahin, A.; Mendez, R.; Dai, J.L. Alternative splicing disrupts a nuclear localization signal in spleen tyrosine kinase that is required for invasion suppression in breast cancer. Cancer Res. 2003, 63, 4724–4730. [Google Scholar]
- Manjili, M.H.; Henderson, R.; Wang, X.Y.; Chen, X.; Li, Y.; Repasky, E.; Kazim, L.; Subjeck, J.R. Development of a recombinant HSP110-HER-2/neu vaccine using the chaperoning properties of HSP110. Cancer Res. 2002, 62, 1737–1742. [Google Scholar]
- Guo, C.; Subjeck, J.R.; Wang, X.Y. Creation of Recombinant Chaperone Vaccine Using Large Heat Shock Protein for Antigen-Targeted Cancer Immunotherapy. Methods Mol. Biol. 2018, 1709, 345–357. [Google Scholar]
- Yasuda, K.; Nakai, A.; Hatayama, T.; Nagata, K. Cloning and expression of murine high molecular mass heat shock proteins, HSP105. J. Biol. Chem. 1995, 270, 29718–29723. [Google Scholar]
- Saito, Y.; Yamagishi, N.; Hatayama, T. Nuclear localization mechanism of Hsp105beta and its possible function in mammalian cells. J. BioChem. 2009, 145, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Olszak, T.; Neves, J.F.; Dowds, C.M.; Baker, K.; Glickman, J.; Davidson, N.O.; Lin, C.S.; Jobin, C.; Brand, S.; Sotlar, K.; et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature 2014, 509, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, N.; Fujii, H.; Saito, Y.; Hatayama, T. Hsp105beta upregulates hsp70 gene expression through signal transducer and activator of transcription-3. FEBS J. 2009, 276, 5870–5880. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Kakunda, M.; Pham, V.; Lill, J.R.; Du, P.; Wongchenko, M.; Yan, Y.; Firestein, R.; Huang, X. HSP105 recruits protein phosphatase 2A to dephosphorylate beta-catenin. Mol. Cell Biol. 2015, 35, 1390–1400. [Google Scholar] [CrossRef]
- Boudesco, C.; Verhoeyen, E.; Martin, L.; Chassagne-Clement, C.; Salmi, L.; Mhaidly, R.; Pangault, C.; Fest, T.; Ramla, S.; Jardin, F.; et al. HSP110 sustains chronic NF-kappaB signaling in activated B-cell diffuse large B-cell lymphoma through MyD88 stabilization. Blood 2018, 132, 510–520. [Google Scholar] [CrossRef]
- Berthenet, K.; Bokhari, A.; Lagrange, A.; Marcion, G.; Boudesco, C.; Causse, S.; De Thonel, A.; Svrcek, M.; Goloudina, A.R.; Dumont, S.; et al. HSP110 promotes colorectal cancer growth through STAT3 activation. Oncogene 2017, 36, 2328–2336. [Google Scholar] [CrossRef]
- Ono, K.; Eguchi, T.; Sogawa, C.; Calderwood, S.K.; Futagawa, J.; Kasai, T.; Seno, M.; Okamoto, K.; Sasaki, A.; Kozaki, K.I. HSP-enriched properties of extracellular vesicles involve survival of metastatic oral cancer cells. J. Cell BioChem. 2018, 119, 7350–7362. [Google Scholar] [CrossRef]
- Eguchi, T.; Sogawa, C.; Okusha, Y.; Uchibe, K.; Iinuma, R.; Ono, K.; Nakano, K.; Murakami, J.; Itoh, M.; Arai, K.; et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. PLoS ONE 2018, 13, e0191109. [Google Scholar] [CrossRef]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef]
- Colgan, S.P.; Pitman, R.S.; Nagaishi, T.; Mizoguchi, A.; Mizoguchi, E.; Mayer, L.F.; Shao, L.; Sartor, R.B.; Subjeck, J.R.; Blumberg, R.S. Intestinal heat shock protein 110 regulates expression of CD1d on intestinal epithelial cells. J. Clin. Investig. 2003, 112, 745–754. [Google Scholar] [CrossRef]
- Berthenet, K.; Boudesco, C.; Collura, A.; Svrcek, M.; Richaud, S.; Hammann, A.; Causse, S.; Yousfi, N.; Wanherdrick, K.; Duplomb, L.; et al. Extracellular HSP110 skews macrophage polarization in colorectal cancer. Oncoimmunology 2016, 5, e1170264. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toft, D.; Reid, J.; Ames, M.; Stensgard, B.; Safgren, S.; Adjei, A.A.; Sloan, J.; Atherton, P.; Vasile, V.; et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Boudesco, C.; Cause, S.; Jego, G.; Garrido, C. Hsp70: A Cancer Target Inside and Outside the Cell. Methods Mol. Biol. 2018, 1709, 371–396. [Google Scholar] [PubMed]
- Eguchi, T.; Lang, B.J.; Murshid, A.; Prince, T.; Gong, J.; Calderwood, S.K. Regulatory Roles for Hsp70 in Cancer Incidence and Tumor Progression. In Role of Molecular Chaperones in Structural Folding, Biological Functions, and Drug Interactions of Client Proteins; Galigniana, M.D., Ed.; Bentham Science Publishers: Sharjah, UAE, 2018; Volume 1, pp. 1–22. [Google Scholar]
- Liu, T.; Daniels, C.K.; Cao, S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Gierasch, L.M. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc. Natl. Acad. Sci. USA 2011, 108, 6987–6992. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Guh, J.Y.; Chen, H.C.; Yang, Y.L.; Huang, J.S.; Chuang, L.Y. Advanced glycation end-product-induced mitogenesis is dependent on Janus kinase 2-induced heat shock protein 70 in normal rat kidney interstitial fibroblast cells. Transl. Res. 2007, 149, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Rao, I.; Earp, J.C.; Jusko, W.J.; Wetzler, M. Down-regulation of heat shock protein 70 improves arsenic trioxide and 17-DMAG effects on constitutive signal transducer and activator of transcription 3 activity. Cancer ChemoTher. Pharmacol. 2010, 66, 681–689. [Google Scholar] [CrossRef]
- Sinn, D.I.; Kim, S.J.; Chu, K.; Jung, K.H.; Lee, S.T.; Song, E.C.; Kim, J.M.; Park, D.K.; Kun Lee, S.; Kim, M.; et al. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. NeuroBiol. Dis. 2007, 26, 464–472. [Google Scholar] [CrossRef]
- Uchida, S.; Fujiki, M.; Nagai, Y.; Abe, T.; Kobayashi, H. Geranylgeranylacetone, a noninvasive heat shock protein inducer, induces protein kinase C and leads to neuroprotection against cerebral infarction in rats. NeuroSci. Lett. 2006, 396, 220–224. [Google Scholar] [CrossRef]
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Arrigo, A.P.; Calderwood, S.K. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Arch. Toxicol. 2013, 87, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Binder, R.J.; Srivastava, P.K. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat. Immunol. 2005, 6, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Menoret, A.; Srivastava, P. Roles of heat-shock proteins in antigen presentation and cross-presentation. Curr. Opin. Immunol. 2002, 14, 45–51. [Google Scholar] [CrossRef]
- Srivastava, P. Interaction of heat shock proteins with peptides and antigen presenting cells: Chaperoning of the innate and adaptive immune responses. Annu. Rev. Immunol. 2002, 20, 395–425. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef]
- Elsner, L.; Muppala, V.; Gehrmann, M.; Lozano, J.; Malzahn, D.; Bickeböller, H.; Brunner, E.; Zientkowska, M.; Herrmann, T.; Walter, L.; et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J. Immunol. 2007, 179, 5523–5533. [Google Scholar] [CrossRef]
- Gross, C.; Hansch, D.; Gastpar, R.; Multhoff, G. Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94. Biol. Chem. 2003, 384, 267–279. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]
- Chen, T.; Guo, J.; Han, C.; Yang, M.; Cao, X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J. Immunol. 2009, 182, 1449–1459. [Google Scholar] [CrossRef]
- Starr, R.; Metcalf, D.; Elefanty, A.G.; Brysha, M.; Willson, T.A.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc. Natl. Acad. Sci. USA 1998, 95, 14395–14399. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, W.W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef]
- Babon, J.J.; Kershaw, N.J.; Murphy, J.M.; Varghese, L.N.; Laktyushin, A.; Young, S.N.; Lucet, I.S.; Norton, R.S.; Nicola, N.A. Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 2012, 36, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.X.; Qian, C.; Liu, B.; Wang, C.; Liu, H.; Pan, X.; Teng, P.; Hu, L.; Zhang, G.; Han, Y.; et al. Induction of suppressor of cytokine signaling 3 via HSF-1-HSP70-TLR4 axis attenuates neuroinflammation and ameliorates postoperative pain. Brain Behav. Immun. 2018, 68, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xiao, G.; Liu, H.; Chen, G.; Wang, X.; Wen, P.; Li, T.; Wen, J.; Xiao, X. Heat Shock Factor 1 Inhibits the Expression of Suppressor of Cytokine Signaling 3 in Cerulein-Induced Acute Pancreatitis. Shock 2018, 50, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Stephanou, A.; Isenberg, D.A.; Nakajima, K.; Latchman, D.S. Signal transducer and activator of transcription-1 and heat shock factor-1 interact and activate the transcription of the Hsp-70 and Hsp-90beta gene promoters. J. Biol. Chem. 1999, 274, 1723–1728. [Google Scholar] [CrossRef]
- Matozaki, M.; Saito, Y.; Yasutake, R.; Munira, S.; Kaibori, Y.; Yukawa, A.; Tada, M.; Nakayama, Y. Involvement of Stat3 phosphorylation in mild heat shock-induced thermotolerance. Exp. Cell Res. 2019, 377, 67–74. [Google Scholar] [CrossRef]
- Saito, Y.; Yamagishi, N.; Ishihara, K.; Hatayama, T. Identification of alpha-tubulin as an hsp105alpha-binding protein by the yeast two-hybrid system. Exp. Cell Res. 2003, 286, 233–240. [Google Scholar] [CrossRef]
- Fritchley, S.J.; Kirby, J.A.; Ali, S. The antagonism of interferon-gamma (IFN-gamma) by heparin: Examination of the blockade of class II MHC antigen and heat shock protein-70 expression. Clin. Exp. Immunol. 2000, 120, 247–252. [Google Scholar] [CrossRef]
- Chen, X.S.; Zhang, Y.; Wang, J.S.; Li, X.Y.; Cheng, X.K.; Wu, N.H.; Shen, Y.F. Diverse effects of Stat1 on the regulation of hsp90alpha gene under heat shock. J. Cell BioChem. 2007, 102, 1059–1066. [Google Scholar] [CrossRef]
- Cheng, M.B.; Zhang, Y.; Zhong, X.; Sutter, B.; Cao, C.Y.; Chen, X.S.; Cheng, X.K.; Xiao, L.; Shen, Y.F. Stat1 mediates an auto-regulation of hsp90beta gene in heat shock response. Cell Signal. 2010, 22, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Pak, S.H.; Joung, Y.H.; Park, J.H.; Lim, E.J.; Darvin, P.; Na, Y.M.; Hong, D.Y.; Lee, B.; Hwang, T.S.; Park, T.; et al. Hypoxia upregulates Hsp90α expression via STAT5b in cancer cells. Int. J. Oncol. 2012, 41, 161–168. [Google Scholar] [PubMed]
- Song, H.; Ethier, S.P.; Dziubinski, M.L.; Lin, J. Stat3 modulates heat shock 27kDa protein expression in breast epithelial cells. Biochem. Biophys. Res. Commun. 2004, 314, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.W.; Chen, Y.; Liu, W.; Chen, Y.J.; Fan, Z.M.; Liu, M.; Li, L.J. Inhibition of JAK2/STAT3 Signaling Pathway Suppresses Proliferation of Burkitt’s Lymphoma Raji Cells via Cell Cycle Progression, Apoptosis, and Oxidative Stress by Modulating HSP70. Med. Sci. Monit. 2018, 24, 6255–6263. [Google Scholar] [CrossRef]
- Krawczyk, Z.; Gogler-Pigłowska, A.; Sojka, D.R.; Scieglinska, D. The Role of Heat Shock Proteins in Cisplatin Resistance. Anticancer Agents Med. Chem. 2018, 18, 2093–2109. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Saini, J.; Sharma, P.K. Clinical, Prognostic and Therapeutic Significance of Heat Shock Proteins in Cancer. Curr. Drug Targets 2018, 19, 1478–1490. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Study Type | Cancer Model | Ref. | |
|---|---|---|---|---|
| Name | Nature/Structure | |||
| Target: HSP27 | ||||
| Apatorsen (OGX-427) | 2nd generation 2’-methoxyethyl-modified ASOs | in vitro/preclinical | Prostate, Ovary | [13,14] |
| clinical trial (phase I) | CRPC, Breast, Ovary, Lung, Bladder | [10] | ||
| in vitro/preclinical | Pancreatic, NSCLC | [15,16] | ||
| clinical trial (phase II) | Stage IV non-squamous NSCLC | [17] | ||
| 3-arylethynyltriazolyl ribonucleoside | ASOs | in vitro | Pancreatic | [18] |
| ASOs-Hsp27 | ASOs | in vitro | Lymphoma | [19] |
| RP101 (Brivudine) | Uridine derivative and nucleoside analog | in vitro/preclinical/clinical | Pancreatic | [20] |
| Target: HSP70 | ||||
| Pifithrin-µ (PFTµ, PES) | Drug-like small molecule | in vitro | AML, ALL, Primary AML blasts | [21,22] |
| VER-155008 | ATP-derivative inhibitor | in vitro | Breast, Colon, Prostatic, Myeloma | [23,24,25] |
| A17/A8 | Peptide aptamer | in vitro/preclinical | Cervix (HeLa cells), Melanoma | [26] |
| ADD70 | Peptide aptamer | in vitro/preclinical | Rat colon carcinoma, Mouse melanoma | [27] |
| cmHsp70.1 | Antibody | preclinical | Colorectal | [28] |
| Hsp70-peptide targeted NK based adoptive immunotherapy | A specific amino acid sequence (TKD) of Hsp70 | clinical trials (phase I/II) | NSCLC (and colon cancer) patients with ex vivo Hsp70 peptide activated, autologous NK | [29] |
| Target: HSP90 | ||||
| Radicicol | natural product isolated from the fungus Monosporium bonorden | in vitro | CML | [30] |
| 17-AAG; 17-DMAG | Derivative of the antibiotic geldanamycin | in vitro/preclinical | Breast, Brain, Medulloblastoma | [31,32,33] |
| 17-DMAG | in vitro | CLL | [34] | |
| IPI-504 (retaspimycin) | Water-soluble derivate of 17-AAG | in vitro/preclinical | Breast, Pancreatic, Metastatic gastrointestinal stromal tumor | [35,36,37,38,39] |
| in vitro/preclinical | NSCLC | [40] | ||
| IPI-504, AUY922 Ganetespib, Onalespib | - | clinical trials (phase I–III) | NSCLC Breast, Ovary, Colon | [41] |
| Novobiocin | Aminocoumarin antibiotic, produced by the actinomycete Streptomyces nivens | in vitro/preclinical | Leukemia, Prostate | [42,43,44] |
| Panaxynol | Natural pesticide and fatty alcohol | in vitro/preclinical | Lung | [45] |
| Ganetespib (STA-9090) | Synthetic, non-geldanamycin, small molecule inhibitor | preclinical | Thyroid | [46] |
| in vitro | Breast | [47] | ||
| BIIB021 (CNF2024) | Orally available, fully synthetic purine scaffold, small molecule inhibitor | in vitro/preclinical | Blood malignancies, Solid tumors | [48] |
| PU-H71 | Non-ansamycin, purine scaffold inhibitor | preclinical | mouse models of the MPN PV and ET | [49] |
| MPN | [50] | |||
| NVP_AUY922 (AUY922) | Esorcinylic isoxazole amide, second-generation non-geldanamycin inhibitor | in vitro/preclinical | Gastric, Small cell lung, Thyroid | [51,52,53,54,55] |
| in vitro | 32D mouse hematopoietic cells expressing wild-type BCR-ABL (b3a2, 32Dp210) and mutant BCR-ABL imatinib-resistant cell lines | [56] | ||
| in vitro/preclinical | Drug-resistant chronic myelogenous leukemia | [57] | ||
| clinical trial (phase II) | Myeloproliferative neoplasms | [58] | ||
| clinical trials (phase I/II) | EGFR-mutant lung cancer with acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors | [59] | ||
| AUY922, HSP990, PU-H71 | - | in vitro/preclinical | Leukemia | [60] |
| Onalespib (AT13387) | second-generation, non-ansamycin inhibitor | in vitro | Transformed kidney cells, primary lung adenocarcinoma | [61] |
| in vitro/preclinical | Melanoma | [62] | ||
| in vitro/preclinical | NSCLC | [63] | ||
| in vitro/preclinical | NSCLC | [64] | ||
| XL888 | Orally available inhibitor with high selectivity for HSP90α and HSP90β | clinical trial (phase I) | Melanoma | [65] |
| SNX2112 SNX5422 | Orally bioavailable, synthetic, small molecule inhibitors that competitively bind to HSP90α, HSP90β, Grp94 and Trap-1 | in vitro/preclinical | Head and neck squamous cell carcinoma | [66] |
| NSCLC | [67] | |||
| CUDC-305, Ganetespib CH5164840, WK88-1 17-DMAG | - | preclinical | NSCLC | [68,69,70,71] |
| Target: HSP110 | ||||
| Foldamers 33 and 52 | Protein–protein interaction inhibitors, based on pyridyl scaffolds mimicking α-helix | in vitro/preclinical | Colorectal | [72] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jego, G.; Hermetet, F.; Girodon, F.; Garrido, C. Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy. Cancers 2020, 12, 21. https://doi.org/10.3390/cancers12010021
Jego G, Hermetet F, Girodon F, Garrido C. Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy. Cancers. 2020; 12(1):21. https://doi.org/10.3390/cancers12010021
Chicago/Turabian StyleJego, Gaëtan, François Hermetet, François Girodon, and Carmen Garrido. 2020. "Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy" Cancers 12, no. 1: 21. https://doi.org/10.3390/cancers12010021
APA StyleJego, G., Hermetet, F., Girodon, F., & Garrido, C. (2020). Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy. Cancers, 12(1), 21. https://doi.org/10.3390/cancers12010021