The MicroRNA-23b/27b/24 Cluster Facilitates Colon Cancer Cell Migration by Targeting FOXP2


Abstract
1. Introduction
2. Results
2.1. Identification of miRNAs Responsible for the High Migration Capacity
2.2. Transcription of Short C9orf3 Isoforms in the MG Cells
2.3. Regulation of Short C9orf3 Expression
2.3.1. Effect of DNA Methylation on C9orf3 and miR-23b/27b/24 Expression
2.3.2. Promoter Array of Short C9orf3 Transcript
2.4. Effects of E2F1 on C9orf3 and miR-23b/27b/24 Expression
2.5. Effects of the miR-23b/27b/24 Cluster on Cell Migration
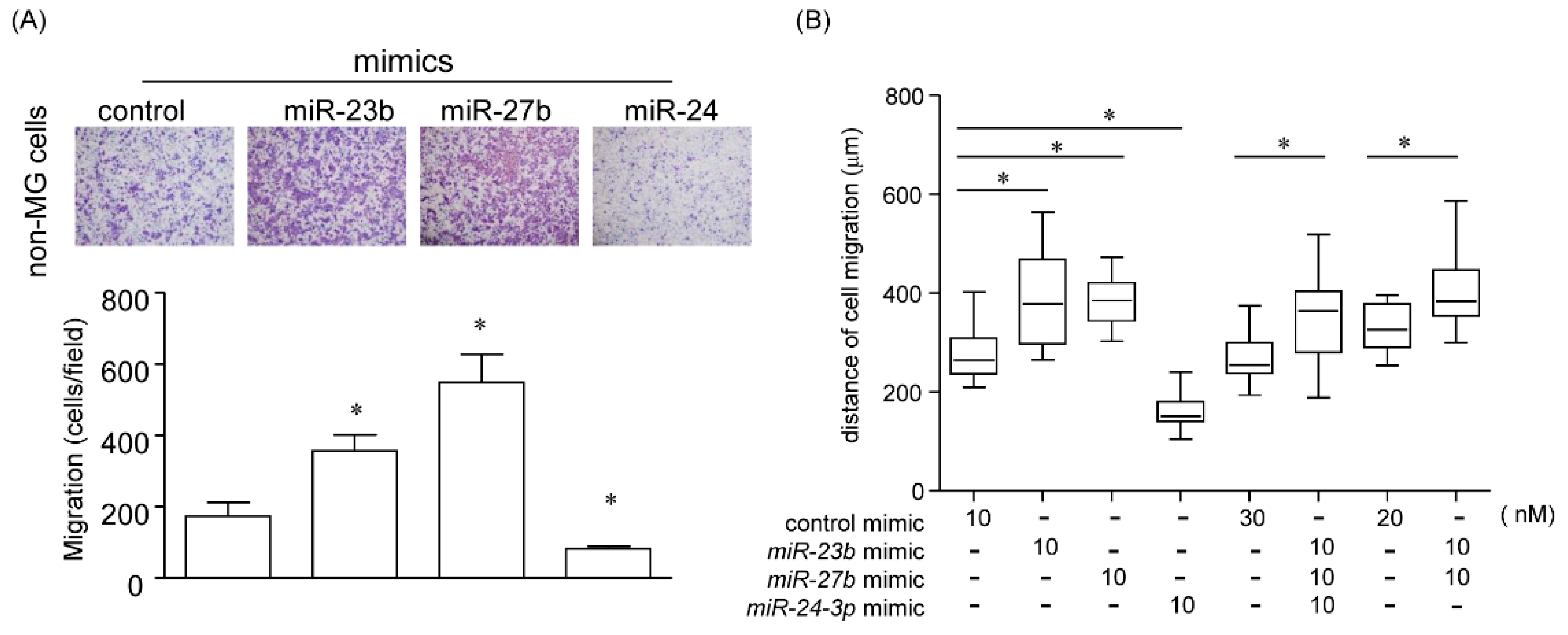
2.5.1. Effect of miR-23b, miR-27b, and miR-24 on Cell Migration
2.5.2. Effect of a Single MicroRNA on Cell Migration
2.6. Identification of Common Target mRNAs of miR-23b and miR-27b
2.7. miR-23b and miR-27b Directly Target the 3′ UTR of FOXP2
2.8. Promotion of Cell Migration in the FOXP2 Knockdown Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Isolation of a Subpopulation with High Cell Migration Capacity
4.2. Cell Counting, Cell Migration Assay and Scratch Assay
4.3. Small Interference RNAs (siRNAs)
4.4. Overexpression of miRNA Mimics and Inhibitors
4.5. Quantitative Real-Time Reverse Transcription-PCR (qPCR)
4.6. Analysis of Global miRNA Expression
4.7. miRNA Luciferase Reporter Assay
4.8. Western Blotting
4.9. Promoter Activity Assay
4.10. Chromatin Immunoprecipitation (ChIP) Assay
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Leva, G.D.; Garofalo, M.; Croce, C.M. microRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar] [CrossRef]
- Xu, J.; Wong, C. A computational screen for mouse signaling pathways targeted by microRNA clusters. RNA 2008, 14, 1276–1283. [Google Scholar] [CrossRef]
- Guo, L.; Lu, Z. Global expression analysis of miRNA gene cluster and family based on isomiRs from deep sequencing data. Comput. Biol. Chem. 2010, 34, 165–171. [Google Scholar] [CrossRef]
- Yu, J.; Wang, F.; Yang, G.-H.; Wang, F.-L.; Ma, Y.-N.; Du, Z.-W.; Zhang, J.-W. Human microRNA clusters: Genomic organization and expression profile in leukemia cell lines. Biochem. Biophys. Res. Commun. 2006, 349, 59–68. [Google Scholar] [CrossRef]
- Ding, L.; Ni, J.; Yang, F.; Huang, L.; Deng, H.; Wu, Y.; Ding, X.; Tang, J. Promising therapeutic role of miR-27b in tumor. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Liu, W.; van Wijnen, A.J. Regulation and biological roles of the multifaceted miRNA-23b (MIR23B). Gene 2018, 642, 103–109. [Google Scholar] [CrossRef]
- Wang, J.Y.; Li, X.F.; Li, P.Z.; Zhang, X.; Xu, Y.; Jin, X. MicroRNA-23b regulates nasopharyngeal carcinoma cell proliferation and metastasis by targeting E-cadherin. Mol. Med. Rep. 2016, 14, 537–543. [Google Scholar] [CrossRef]
- Yao, J.; Deng, B.; Zheng, L.; Dou, L.; Guo, Y.; Guo, K. MiR-27b is upregulated in cervical carcinogenesis and promotes cell growth and invasion by regulating CDH11 and epithelial-mesenchymal transition. Oncol. Rep. 2016, 35, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wessely, O.; Marcusson, E.G.; Ivan, C.; Calin, G.A.; Alahari, S.K. Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-A in breast cancer. Cancer Res. 2013, 73, 2884–2896. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kuwano, Y.; Nishikawa, T.; Rokutan, K. ZNF350 promoter methylation accelerates colon cancer cell migration. Oncotarget 2018, 9, 36750–36769. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef] [PubMed]
- Hovestadt, V.; Jones, D.T.W.W.; Picelli, S.; Wang, W.; Kool, M.; Northcott, P.A.; Sultan, M.; Stachurski, K.; Ryzhova, M.; Warnatz, H.-J.J.; et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 2014, 510, 537–541. [Google Scholar] [CrossRef]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef]
- Messeguer, X.; Escudero, R.; Farre, D.; Nunez, O.; Martinez, J.; Alba, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]
- Liang, Y.X.; Lu, J.M.; Mo, R.J.; He, H.C.; Xie, J.; Jiang, F.N.; Lin, Z.Y.; Chen, Y.R.; Wu, Y.D.; Luo, H.W.; et al. E2F1 promotes tumor cell invasion and migration through regulating CD147 in prostate cancer. Int. J. Oncol. 2016, 48, 1650–1658. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, X.; Bao, Y.; Mo, J.; Du, H.; Hu, J.; Zhang, X. E2F1 silencing inhibits migration and invasion of osteosarcoma cells via regulating DDR1 expression. Int. J. Oncol. 2017, 51, 1639–1650. [Google Scholar] [CrossRef]
- Fang, Z.; Gong, C.; Liu, H.; Zhang, X.; Mei, L.; Song, M.; Qiu, L.; Luo, S.; Zhu, Z.; Zhang, R.; et al. E2F1 promote the aggressiveness of human colorectal cancer by activating the ribonucleotide reductase small subunit M2. Biochem. Biophys. Res. Commun. 2015, 464, 407–415. [Google Scholar] [CrossRef]
- Diao, H.; Ye, Z.; Qin, R. miR-23a acts as an oncogene in pancreatic carcinoma by targeting FOXP2. J. Investig. Med. 2017. [Google Scholar] [CrossRef]
- Zhong, C.; Liu, J.; Zhang, Y.; Luo, J.; Zheng, J. MicroRNA-139 inhibits the proliferation and migration of osteosarcoma cells via targeting forkhead-box P2. Life Sci. 2017, 191, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Cuiffo, B.G.; Campagne, A.; Bell, G.W.; Lembo, A.; Orso, F.; Lien, E.C.; Bhasin, M.K.; Raimo, M.; Hanson, S.E.; Marusyk, A.; et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell 2014, 15, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.-Z.; Yu, T.; An, Q.; Yang, H.; Zhang, Z.; Liu, X.; Xiao, G. MicroRNA-190 regulates FOXP2 genes in human gastric cancer. Onco Targets Ther. 2016, 9, 3643–3651. [Google Scholar] [PubMed]
- Sato, S.; Rancourt, A.; Sato, Y.; Satoh, M.S. Single-cell lineage tracking analysis reveals that an established cell line comprises putative cancer stem cells and their heterogeneous progeny. Sci. Rep. 2016, 6, 23328. [Google Scholar] [CrossRef]
- Mohammadi, A.; Mansoori, B.; Baradaran, B. The role of microRNAs in colorectal cancer. Biomed. Pharmacother. 2016, 84, 705–713. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, J.; Zhang, H.; Lu, J. MicroRNAs in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Mol. Biol. Evol. 2016, 33, 2232–2247. [Google Scholar] [CrossRef]
- Goto, Y.; Kojima, S.; Nishikawa, R.; Enokida, H.; Chiyomaru, T.; Kinoshita, T.; Nakagawa, M.; Naya, Y.; Ichikawa, T.; Seki, N.; et al. The microRNA-23b/27b/24-1 cluster is a disease progression marker and tumor suppressor in prostate cancer. Oncotarget 2014, 5, 7748–7759. [Google Scholar] [PubMed]
- Ell, B.; Qiu, Q.; Wei, Y.; Mercatali, L.; Ibrahim, T.; Amadori, D.; Kang, Y. The microRNA-23b/27b/24 cluster promotes breast cancer lung metastasis by targeting metastasis-suppressive gene prosaposin. J. Biol. Chem. 2014, 289, 21888–21895. [Google Scholar] [CrossRef]
- Lutter, D.; Marr, C.; Krumsiek, J.; Lang, E.W.; Theis, F.J. Intronic microRNAs support their host genes by mediating synergistic and antagonistic regulatory effects. BMC Genom. 2010, 11, 224. [Google Scholar] [CrossRef]
- Diaz-Perales, A.; Quesada, V.; Sanchez, L.M.; Ugalde, A.P.; Suarez, M.F.; Fueyo, A.; Lopez-Otin, C. Identification of human aminopeptidase O, a novel metalloprotease with structural similarity to aminopeptidase B and leukotriene A4 hydrolase. J. Biol. Chem. 2005, 280, 14310–14317. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, D. E2F1 pathways to apoptosis. FEBS Lett. 2002, 529, 122–125. [Google Scholar] [CrossRef]
- Poppy Roworth, A.; Ghari, F.; La Thangue, N.B. To live or let die—Complexity within the E2F1 pathway. Mol. Cell. Oncol. 2015, 2, e970480. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The MicroRNA-17-92 Family of MicroRNA Clusters in Development and Disease. Cancer J. 2012, 18, 262–267. [Google Scholar] [CrossRef]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted Deletion Reveals Essential and Overlapping Functions of the miR-17∼92 Family of miRNA Clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Bender, S.; Raddatz, G.; Ansari, I.; Cohen, D.; Gutekunst, J.; Musch, T.; Linhart, H.; Breiling, A.; Pikarsky, E.; et al. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Res. 2015, 75, 2120–2130. [Google Scholar] [CrossRef]
- Boyden, S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962, 115, 453–466. [Google Scholar] [CrossRef]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. BiorXiv 2019. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Change Relative to Upper Cells (1) (Fold-Change) | mirBase Accession No. |
|---|---|---|
| hsa-miR-10a | 2.119 | MIMAT0000415 |
| hsa-miR-23b | 2.610 | MIMAT0000418 |
| hsa-miR-27b | 1.837 | MIMAT0000419 |
| has-miR-1274b(2) | 1.511 | MIMAT0005938 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, K.; Kuwano, Y.; Rokutan, K. The MicroRNA-23b/27b/24 Cluster Facilitates Colon Cancer Cell Migration by Targeting FOXP2. Cancers 2020, 12, 174. https://doi.org/10.3390/cancers12010174
Nishida K, Kuwano Y, Rokutan K. The MicroRNA-23b/27b/24 Cluster Facilitates Colon Cancer Cell Migration by Targeting FOXP2. Cancers. 2020; 12(1):174. https://doi.org/10.3390/cancers12010174
Chicago/Turabian StyleNishida, Kensei, Yuki Kuwano, and Kazuhito Rokutan. 2020. "The MicroRNA-23b/27b/24 Cluster Facilitates Colon Cancer Cell Migration by Targeting FOXP2" Cancers 12, no. 1: 174. https://doi.org/10.3390/cancers12010174
APA StyleNishida, K., Kuwano, Y., & Rokutan, K. (2020). The MicroRNA-23b/27b/24 Cluster Facilitates Colon Cancer Cell Migration by Targeting FOXP2. Cancers, 12(1), 174. https://doi.org/10.3390/cancers12010174