1. Introduction
Breast cancer is the most common cancer in women [
1] and the second leading cause of global cancer death after lung cancer [
2]. Triple-negative breast cancer (TNBC) represents almost 15–20% of breast cancers [
3] which can be distinguished by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) on breast cancer cells [
1,
4].
TNBC is associated with a higher mortality rate due to its heterogeneous and extremely aggressive nature with limited treatment responses, enhanced recurrence, and metastasis, especially in the lungs and brain [
1,
5,
6]. Conventional single mono-chemotherapy is inadequate for treating TNBC, whereas cyclophosphamide (CYP) is highly reported as a neoadjuvant therapy with an anthracycline, microtubule stabilizers, or platinum agents to achieve improved efficacy and maximum benefits [
6,
7,
8]. However, in these therapies, CYP was administered to the patients in a high dose of almost 600 mg/m
2 [
8] to exert its clinical efficacy [
9,
10], leading to nausea, vomiting, neutropenia [
8], and cardiac toxicity [
11].
The alkylating agent CYP is a prodrug which is metabolized in the liver by cytochrome P450, producing pharmacologically stable toxic metabolites—acrolein and phosphoramide mustard. These metabolites passively enter into the cell and attach to the guanine base of DNA, which hinders DNA replication by establishing intrastrand and interstrand DNA cross-linking [
11,
12]. CYP is a widely used chemotherapeutic in cancer treatment due to its direct cytotoxic effect on cancer cells with an immunoregulatory response [
13] depending on the dose of the drug, as shown in a mouse model, ranging from 20–200 mg/kg [
14,
15,
16].
The main objective of choosing and designing these chemical compounds as chemotherapeutic drugs which are toxic in nature is to destroy all rapidly dividing cells and restrict the spread of cancer. The key shortcoming of this approach is that it not only targets uncontrolled cancer cell proliferation, but also kills the body’s rapidly proliferating normal cells, such as hair follicles and intestinal epithelia, leaving the patient to cope with life-altering side effects [
17,
18]. This downside of the treatment urged the development of smartly designed nanoparticles (NPs) for targeted delivery of anticancer drugs to the tumor site, while avoiding the normal healthy tissues and organs [
19,
20].
NP-mediated anticancer drug delivery systems currently focus on improving the effectiveness of the chemotherapeutics, by altering the pharmacokinetics of the drugs, delivering neoadjuvant therapies, selectively targeting tumor cells, enhancing the cellular uptake of anticancer agents, releasing the payload inside the tumor microenvironment, and simultaneously ensuring the drug delivery and efficacy [
21]. The larger size of NPs prohibits the carrier from crossing the tight junctions of healthy blood vessels; however, the NPs can breach the inflamed or injured tumor site due to leaky junctions, and they can stay in the tumor region due to the absence of lymphatic drainage, allowing the NPs to deliver the anticancer drug in the cancer cell for a prolonged period of time. This phenomenon is known as the enhanced permeability and retention (EPR) effect that could be harnessed for passive targeting [
22].
Among the wide variety of NPs, carbonate apatite (CA) NPs and surface-modified CA NPs attracted increasing attention due to their auspicious therapeutic outcomes in nucleic acid and anticancer drug delivery by virtue of a high drug/nucleic acid encapsulation capacity, favorable pharmacokinetics, enhanced cellular uptake, pH-sensitive payload release, efficient transgene expression or gene silencing, and eventually a significant tumor-inhibitory effect both in vitro [
23,
24,
25,
26,
27,
28] and in vivo [
29,
30,
31,
32,
33,
34,
35].
CA NPs possess PO
43−/HCO
3−- and Ca
2+-rich domains on their outer surface. Due to the heterogeneous surface charge, these NPs provide an ample opportunity to bind drugs via ionic interactions. The drug-loaded CA NPs cross the cell membrane from systemic circulation through an energy-dependent process termed as endocytosis. After the internalization of the cargo, it encounters a highly acidic environment inside the endosomal compartment where the phosphate and carbonate ions in the CA structure tend to consume excess protons from the microenvironment, leading to low pH-dependent dissolution of NPs and consequent drug release [
36,
37,
38].
Previously, we successfully synthesized and characterized CA NPs, citrate-modified CA (CMCA) NPs [
28,
39], succinate-modified CA (SMCA) NPs, and α-ketoglutaric acid-modified CA (α-KAMCA) NPs [
40], and we evaluated their particle size, drug-loading efficiency, enhancement in cytotoxicity, and cell viability in a human breast cancer cell line (MCF-7 cells) and a murine breast cancer cell line (4T1 cells). Here, we examined CA, CMCA, and α-KAMCA NPs for in vivo tumor regression, biodistribution, plasma concentration, and toxicology study depending on their promising in vitro results, especially through a cytotoxicity assay in the 4T1 cell line. These 4T1 cells are from an extremely tumorigenic and aggressive mouse mammary adenocarcinoma cell line, resembling many characteristics of human TNBC [
41], and they represent an in vivo model for human stage IV breast cancer.
Doxorubicin (DOX) is an anthracycline group chemotherapeutic that prevents DNA replication by breaking down DNA chains during DNA supercoiling, and it also produces free radicals that induce DNA and cell membrane damage. However, DOX causes major side effects such as cardiomyopathy, leading to congestive heart failure and death [
42,
43,
44,
45]. In our study, we loaded DOX into CA, CMCA, and α-KAMCA NPs owing to its fluorescent properties, prior to tracking accumulation and distribution of the drug-loaded cargos in different tissues, tumor, and plasma by using fluorescence-based techniques. For the tumor regression study, CYP was incorporated into CA, CMCA, and α-KAMCA NPs, and the NPs without drug were also subjected to a toxicology study to analyze any potential side effects caused by the NPs on the healthy tissues.
3. Discussion
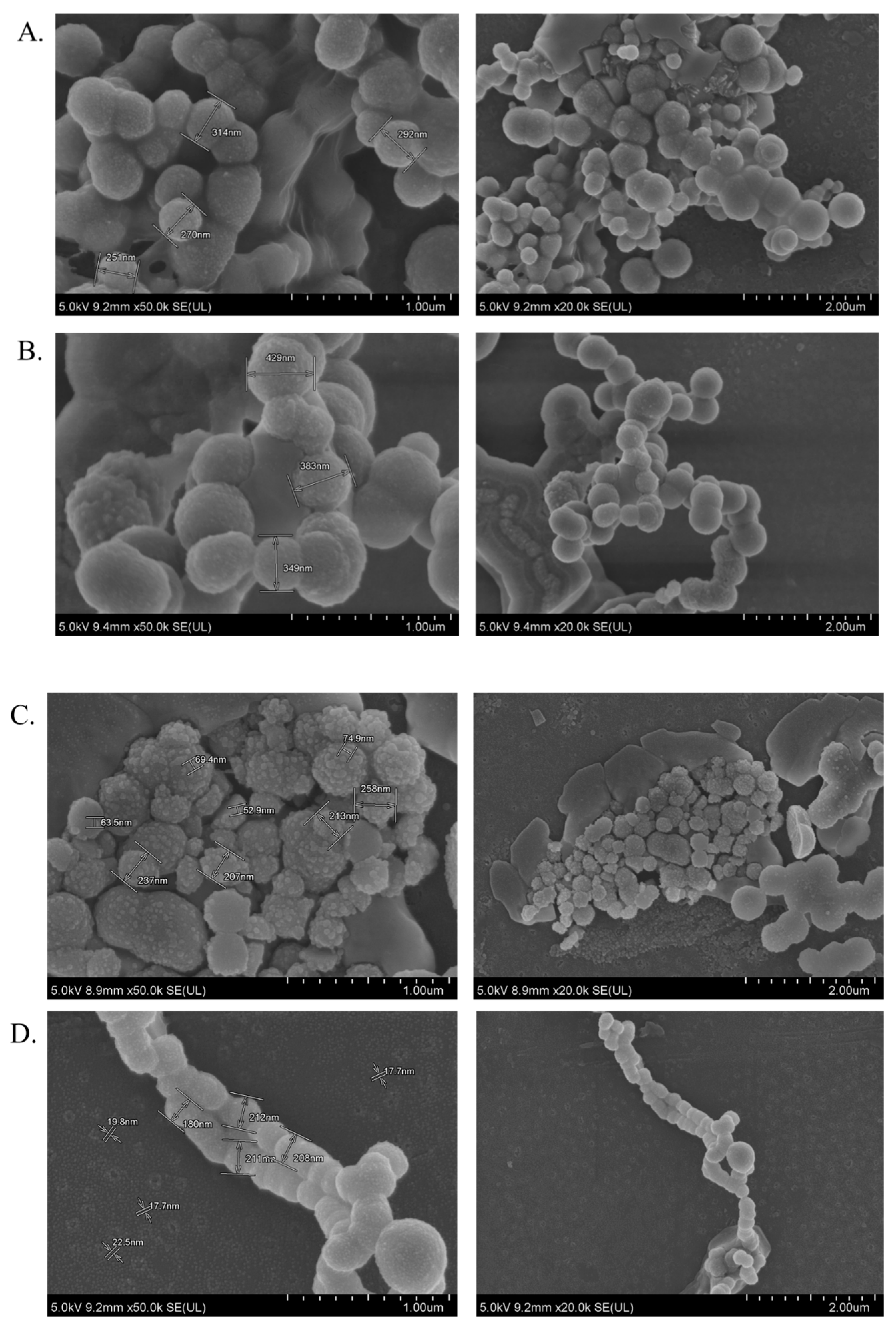
The average particle size/diameter with its polydispersity index (PDI) is a determinant to control the maximum cellular uptake, internalization, biodistribution, drug release profile, and bioavailability of an encapsulated therapeutic compound [
54,
55,
56]. CA NPs, CMCA NPs, and α-KAMCA NPs prepared using a salt precipitation technique resulted in smaller particle sizes which were, respectively, 428.4 ± 21.70 nm, 163 ± 10.96 nm, and 291.5 ± 10.60 nm [
28,
39]. Drug-loaded NPs showed a particle size ranging from 180–436 nm (
Figure 9). In our previous study [
28,
39,
40], we recorded the surface charges for all NP formulations within the range of −9 to −13 mV with a PDI value of 0.553, 0.787, and 0.322 for CA, CMCA, and α-KAMCA NPs, respectively. The PDI values of the nanocarriers pointed out that α-KAMCA NPs are homogeneous (monodisperse) compared to CA and CMCA NPs. The size and PDI value together play pivotal roles in influencing the endocytosis-dependent cellular uptake, especially for in vivo applications.
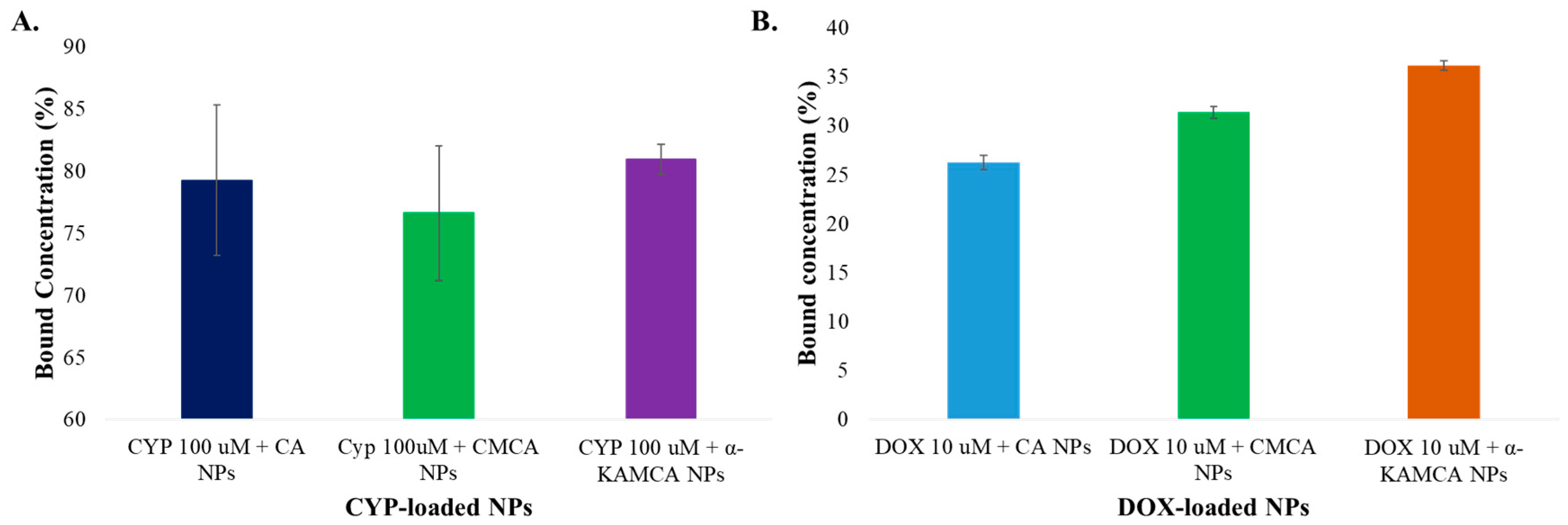
In contrast, the drug-loading capacity or binding affinity of the NPs depends on the solubility profile of the drug and also the properties of the carrier molecules which involve molecular weight, chemical structure, drug–salt interaction, and the presence of functional groups [
22]. CYP showed higher binding affinity ranging from 76–81% toward CA, CMCA, and α-KAMCA NPs at physiological pH (7.4), indicating that the protonated secondary and tertiary cationic amine groups of the CYP structure might bind to the anionic domains of the nanoparticles via electrostatic interaction. CMCA, SMCA (succinate-modified carbonate apatite), and α-KAMCA NPs were prepared by modifying CA with citrate, containing three carboxyl groups, succinate, containing two carboxyl groups, and ketoglutarate, containing one ketone group and two carboxylic groups, respectively. In our previous experiment [
28,
39,
40], we showed that the presence of carboxylic and ketone groups in the chemical structure has an influential role in determining the particle size and the binding affinity of the drugs toward the NPs. The α-KAMCA NPs exhibited the desired particle size with a large number of resultant particles and a folded larger surface morphology with rapid dissolution at acidic pH in the microenvironment, resulting in more significant cytotoxicity than the CA NPs.
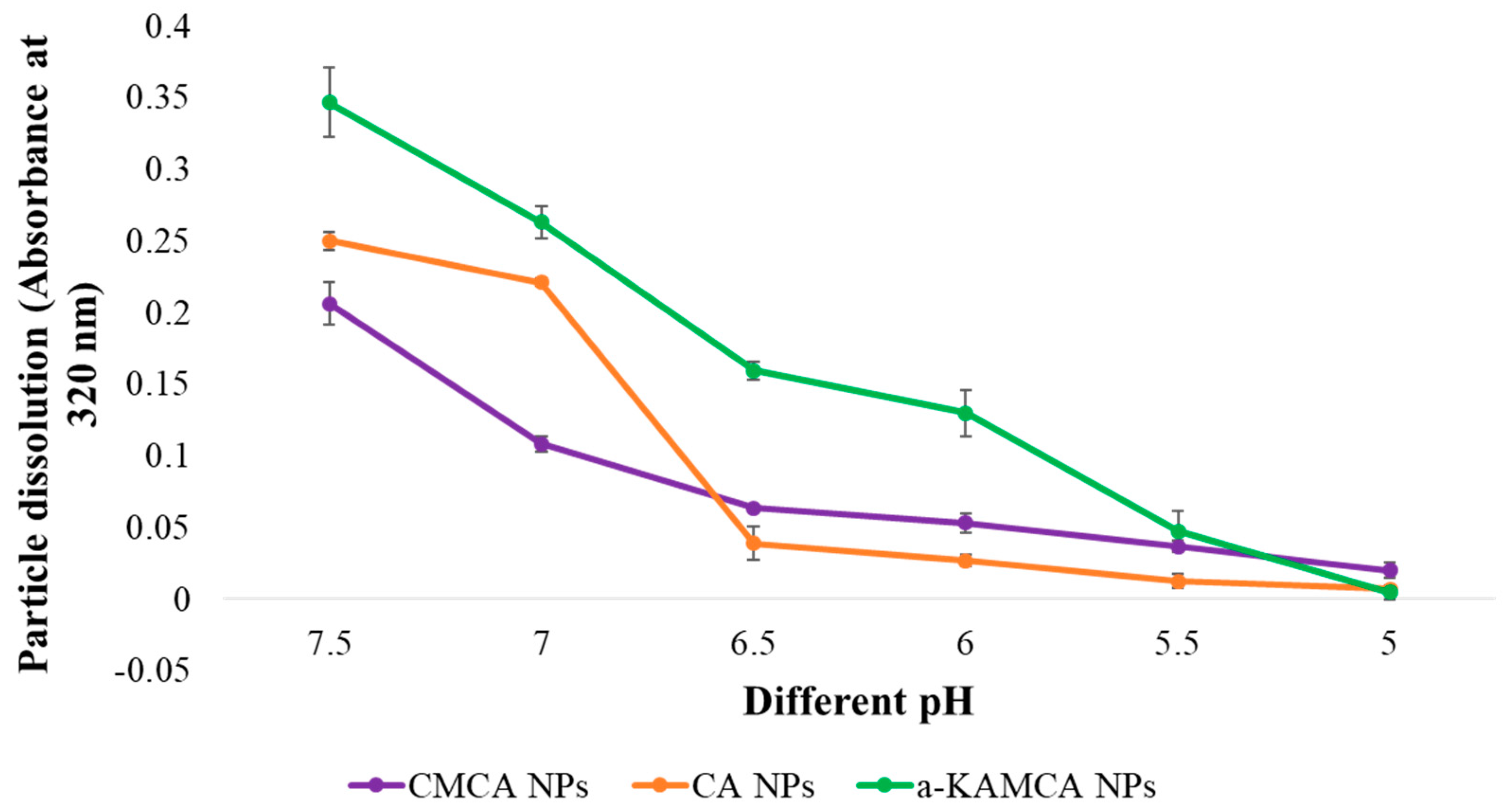
The targeted delivery of the drug–NP complex to the specific tissue or organ could be achieved by modifying the size, surface charge, and the surface morphology of the NPs. However, there would be no meaningful outcome if the drug could not be released from the nanoparticle matrix. The release of drug from the nanoparticle-based formulation depends on many factors including pH, temperature, drug solubility, desorption of the surface-bound or adsorbed drug, drug diffusion through the nanoparticle matrix, nanoparticle matrix swelling and erosion, and the combination of erosion and diffusion processes [
57,
58,
59]. The influence of the pH-sensitive dissolution of CA, CMCA, and α-KAMCA NPs ensured drug release in a acidic microenvironment [
28,
40] in our former study. An in vitro turbidity test (
Figure 11) was conducted by modeling physiological pH (7.4) and endosomal acidic pH (6.5–5.0), and the NPs were subjected to these environments to ensure pH sensitivity. The NPs were stable at physiological blood pH (7.4), but rapidly dissolved in the weakly acidic microenvironment. Moreover, all of the NPs released their payload within 5 min at late endosomal pH (pH 5), suggesting possible discharge of the drug into the cytoplasm. The phosphate (PO
43−) and carbonate ions (CO
32−) present in the apatite structure could readily accept excess H
+ ions from the endosomal acidic microenvironment, causing the particles to be dissolved [
38]. Furthermore, the cation (Ca
2+) and anions (PO
43−, CO
32−) released from the particles might create osmotic pressure across the endosomal membrane, which in turn could lead to endosome swelling and rupture, releasing the payload into the cytoplasm [
23,
60]. However, after the dissolution of the NPs in the endosome, the drug might also enter into the cytoplasm via passive diffusion through the endosomal membrane.
Intravenous administration of NPs is the most common route as it can bypass the epithelium absorption barrier by directly entering into systemic circulation [
61]. Nanoparticle–protein interaction takes place in circulation prior to distribution into various organs [
48,
62]. As a result, to deliver the drug, NPs require a perfect targeting device to ensure selective accumulation of the drug in the targeted site while maintaining a low concentration in the healthy organs. The size, surface charge, and morphology of the NPs play an important role in targeting and clearance from the body through the mononuclear phagocytic system (MPS), renal system, and the immune system; however, it is important for the NPs to be eliminated from the body within a predetermined time frame to avoid possible systemic toxicity [
48,
63,
64].
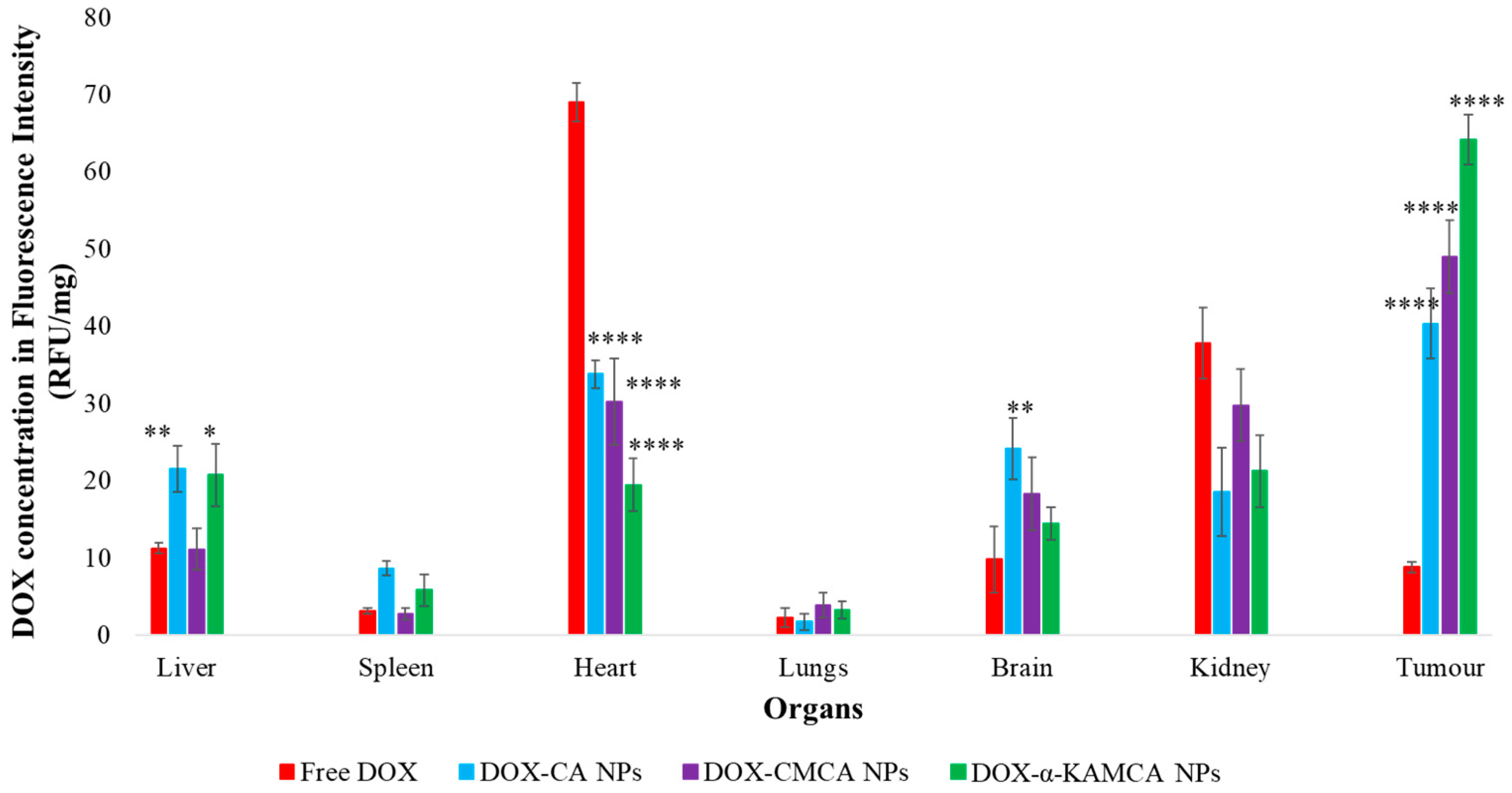
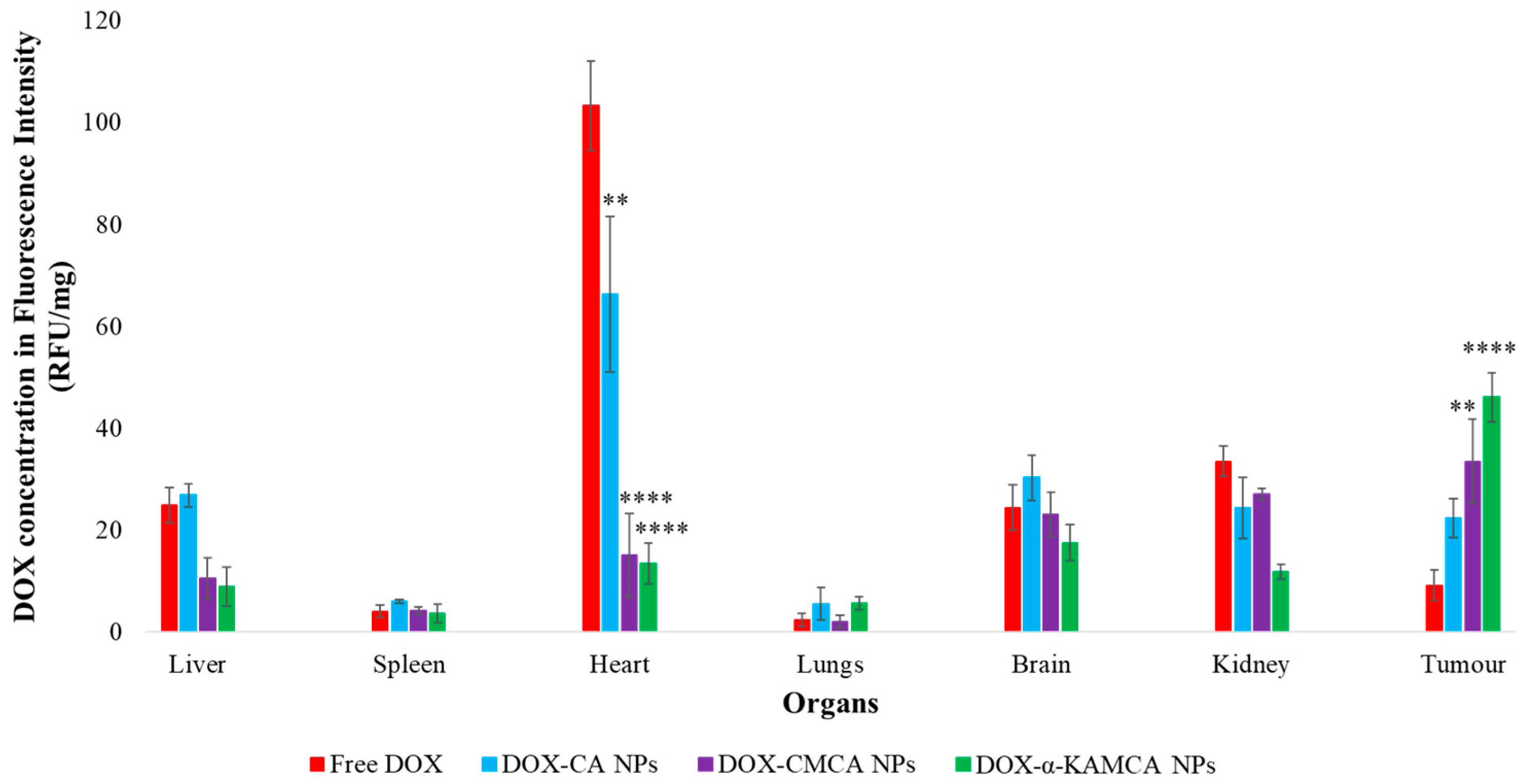
The experimental results reveal that the biodistribution of DOX is greatly altered when delivered through NPs. The NPs significantly enhanced the circulation half-life of DOX in the blood (
Figure 6,
Supplementary material Table S3). Among all types of NPs, at 2 h of treatment, DOX-loaded CMCA NPs showed less liver uptake, which could be due to their rapid renal clearance due to their smaller particle size [
65], ranging from 36 nm to 72 nm (
Figure 7,
Supplementary material Table S1). The α-KAMCA NPs resulted in higher DOX concentrations in blood serum than CA and CMCA NPs, demonstrating their long circulating characteristics. This is further supported by the low distribution of DOX in kidney delivered through α-KAMCA NPs after 24 h. The size of α-KAMCA NPs ranging from 230 nm to 360 nm reduced the probability of renal clearance, with surface-anchored α-KA circumventing opsonization and uptake by the reticuloendothelial system (RES), resulting in a prolonged circulation time for the drug. A very important aspect is the significant reduction in the distribution of DOX to the heart by CA, CMCA, and α-KAMCA NPs, indicating their potential in reducing the cardiotoxicity associated with DOX therapy.
A substantially lower amount of DOX-loaded CA, CMCA, and α-KAMCA NPs were detected in the kidneys compared to free DOX, which could be due to the enhancement of drug retention in plasma by NPs. The cutoff size for glomerular clearance of NPs is ~5 nm [
66], indicating that free drug and very small particles used the kidney as an elimination route. We observed minimal accumulation in the major reticuloendothelial organs, including the lungs and spleen, which means that the NPs were masked from the RES.
Interestingly, the tumor accumulation of DOX was proportionate to the serum DOX level, while almost eight-fold more DOX accumulation was observed for DOX-loaded α-KAMCA NPs than free DOX at 2 h of treatment. Likewise, DOX-loaded CA NPs also showed a proportionate accumulation of drug in the tumor and blood serum, which exhibited almost five-fold more drug accumulation compared to the free form at 2 h of treatment. A similar finding was observed at 24 h of treatment for the same NPs. However, the free DOX accumulation in the heart was 4.28 ± 0.63 ng/mg, which was reduced to 2.73 ± 0.38 ng/mg, 0.59 ± 0.08 ng/mg, and 0.58 ± 0.02 ng/mg by incorporating DOX into CA, CMCA, and α-KAMCA NPs, respectively, at 24 h of treatment, shedding light on the potential application of CMCA and α-KAMCA NPs in avoiding DOX-associated cardiomyopathy. NPs with a diameter range from 100–150 nm have a tendency to circulate in the normal blood vessels and perfuse the capillaries of kidney, lung, and heart tissues [
56,
67]. In this study, the drug-loaded NPs possessed a size between 180 and 436 nm (
Figure 9), which is bigger than the cut-off size for perfusing capillary tissues, especially in the heart, ensuring less accumulation of DOX-loaded NPs in the heart. However, the DOX–nanocarrier complex can easily pass through the leaky vasculature of the tumor region due to the bigger pore size of the tumor vessels, and it can accumulate for a longer period of time by virtue of the impaired lymphatic drainage in the tumor site (EPR effect), resulting in efficient uptake of drug-loaded NPs by target cancer cells with more antitumor efficacy and eventually less cytotoxicity.
The acidic microenvironment surrounding cancer cells contains specific types of proteins which might modify the composition of the protein corona around NPs, resulting in a change in bioavailability, as well as alteration of the interactions between the NPs and membranes of macrophages, endothelial cells, and target cells, and the overall therapeutic response [
68]. The protein–NP interaction influences opsonization, circulation half-life, and cellular uptake. The protein corona can be characterized by total quantity, density, thickness, composition, relative abundance of each protein, protein binding affinity, and protein conformation. Complement factors, fibrinogen, or immunoglobulin (IgG), which are known as opsonins, would induce macrophage recognition and engulfment of NPs, leading to the fast clearance of NPs out of the body. In contrast, albumin or apolipoproteins (Apos), named dysopsonins, could extend the circulation time of the NPs [
69,
70,
71,
72]. This suggests that drug-loaded CA, CMCA, and α-KAMCA NPs are less likely to be cleared out by the reticuloendothelial system and, thus, would have an increased half-life in plasma, which can be further supported by the pharmacokinetics study of NP-treated mice. The α-KAMCA NPs resulted in higher DOX concentrations in blood serum than CA and CMCA NPs, demonstrating their long circulating characteristics due to the presence of dysopsonins on their protein corona. The size of α-KAMCA NPs ranging from 230 nm to 360 nm reduced the probability of reticuloendothelial system (RES) uptake via opsonization, resulting in a prolonged circulation time for the drug. The long circulation property of α-KAMCA NPs further enhanced DOX accumulation in the tumor, supported by the notion that NPs could enter into tumors through an EPR effect.
However, there are some limitations to using DOX fluorescence as a measure of concentration due to its self-quenching or the pH sensitivity of excitation or emission spectra [
73], which was carefully taken care of during our experiment and data interpretation. Firstly, as our NPs are pH-sensitive, the pH of the lysis buffer was strictly maintained at 7.4 during the experiment to maintain consistency. Secondly, in our previous study, we used visualization by confocal microscopy to observe cellular uptake, as well as a cell viability test using different DOX-loaded NPs to correlate between the cellular uptake and cytotoxicity [
28]. Considering all results, we found DOX as a potential model drug to measure fluorescence intensity in different organs and plasma to measure the concentration of free DOX and DOX-loaded NPs.
No definite signs of toxicity in mice treated with CA NPs, CMCA NPs, and α-KAMCA NPs were observed in acute and sub-chronic toxicity analysis (
Table 4 and
Table 5), which could be supported by the tumor regression study. The drug-free NPs showed no difference in tumor proliferation compared to the untreated mice group, pointing out that NPs are safe as a drug carrier without any toxicity.
4. Methods and Materials
4.1. Reagents
Dulbecco’s modified Eagle medium (DMEM) powder was purchased from Gibco by Life Technology (Thermo Fisher Scientific, USA). Calcium chloride dihydrate (CaCl2∙2H2O), sodium bicarbonate (NaHCO3), sodium hydrogen phosphate (Na2HPO4), potassium dihydrogen phosphate (KH2PO4), sodium citrate tribasic dihydrate, and alpha-ketoglutaric acid disodium salt hydrate were obtained from Sigma-Aldrich (St Louis, MO, USA). Anticancer drugs DOX and CYP were acquired from Sigma-Aldrich (St Louis, MO, USA). DMEM liquid media, fetal bovine serum (FBS), trypsin, TrypLE Express, penicillin–streptomycin, and trypan blue were procured from Sigma-Aldrich (St Louis, MO, USA). Sodium chloride and potassium chloride salts were bought from Fischer Scientific (Loughborough, UK). Acetonitrile (ACN), hydrochloric acid (HCl), and methanol were from Fischer Scientific (Loughborough, UK). Ammonium bicarbonate, formic acid, dithiothreitol (DTT), trifluoroacetic (TFA), trifluoroethanol (TFE) acid, and iodoacetamide (IAM) were from Sigma-Aldrich (St. Louis, MO, USA).
4.2. Synthesis and Encapsulation Efficiency of Drug-Loaded CA, CMCA, and α-KAMCA NPs
DMEM-buffered medium was prepared by mixing 0.675 g of DMEM powder with 44 mM sodium bicarbonate in Milli Q water, and the pH of the media was adjusted to 7.4 by using 0.1 M HCl. The final medium was filtered by using a sterilized non-pyrogenic 0.2-µm PES (polyethersulfone) membrane containing a syringe filter (Sartorius Stedim Biotech, Göttingen, Germany).
High-performance liquid chromatography (HPLC) was performed to assess CYP binding affinity with the NPs. CYP at 100 µM (26.11 µg/mL) concentration was added with 4 mM exogenous calcium to 1 mL of filtered DMEM medium to synthesize CYP-loaded CA NPs. Likewise, the same concentration of CYP was used along with 4 mM exogenous Ca2+ and 1 mM sodium citrate in 1 mL of filtered DMEM solution to generate CYP-bound CMCA NPs. To produce CYP-incorporated α-KAMCA NPs, CYP at 100 µM concentration was mixed with 4 mM exogenous Ca2+ and 4 mM alpha-ketoglutaric acid salt in 1 mL of filtered DMEM solution. All the salts and the drug mixture were then placed inside an incubator at 37 °C for 30 min to formulate drug-bound NPs. Next, the drug–particle suspension was centrifuged at 4 °C temperature for 30 min at 13,000 rpm to accelerate the particle sedimentation. The clear supernatant of the sedimented suspension was collected and run by using Agilent Chemostation software attached to HPLC (Agilent, Santa Clara, CA, USA) to check the amount of unbound drug present in the supernatant. This chromatography experiment was performed using a Zorbax C18 column (4.6 × 150 mm, Agilent, Santa Clara, CA, USA), and the mobile phase was ACN and Milli Q water in a proportion of 30:70 (v/v). The run time, diode array detection (DAD) wavelength, injection volume, and column temperature were fixed to 10 min, 197 nm, 20 µL, and 30 °C, respectively.
The standard curve was prepared by dissolving different concentrations of CYP from 0 µM to 100 µM (
y = 7.9458
x + 5.2171,
R2 = 0.9997) to calculate the concentrations of unbound CYP in the supernatant.
The percentage drug encapsulation efficiency was calculated using the following formula:
where
N is the concentration of unbound drug or the volume of drug not entrapped by the NPs,
P is the peak area of CYP present in the supernatant detected by HPLC,
Q is the encapsulated concentration of CYP in the NPs, and
M is the initial or total concentration of CYP used to formulate the CYP-bound NP formulations.
A standard curve was made by plotting different concentrations of DOX (0–15 µM) vs. fluorescence intensity. DOX at 10 µM (5.77 µg/mL) was added to CA NPs, CMCA NPs, and α-KAMCA NPs. After incubation for 30 min at 37 °C to form DOX-loaded CA, CMCA, and α-KAMCA NPs, the particle suspension was centrifuged twice at 6000 rpm for 20 min at 4 °C. Finally, after removing supernatant, the pellet was mixed with 10 mM ethylenediaminetetraacetic acid (EDTA) in phosphate-buffered saline (PBS) to dissolve the particles and release the bound drugs, and it was then subjected to a fluorescence intensity measurement with an excitation wavelength of 485 nm and an emission wavelength of 535 nm using Perkin Elmer 2030 manager software attached to a 2030 multilabel reader victor X5 (Perkin Elmer).
The concentrations of DOX present in the suspension were calculated from the fluorescence intensity, using the standard curve. The percentage binding efficiency was calculated using the following formula:
where [
Z]
NP bound drug is the fluorescence intensity of DOX-loaded NPs, and [
Z]
initial is the fluorescence intensity of DOX used to prepare the standard curve. The experiments were performed in triplicate, and the results were presented as means ± SD (standard deviation).
4.3. Morphology Analysis of CYP- and DOX-Loaded CA and α-KAMCA NPs by FE-SEM
CYP or DOX in 1 µM concentration was added along with 4 mM exogenous calcium and/or alpha-ketoglutaric acid and/or sodium citrate salt to 1 mL of filtered DMEM medium prior to incubation at 37 °C for 30 min to formulate CYP- or DOX-loaded NPs. The formulated drug-loaded NPs were centrifuged at 13,000 rpm for 15 min with a refrigerated bench-top microcentrifuge to collect the pellets. Then, 10 μL of each sample was withdrawn and placed onto a carbon tape-coated sample holder and dried at room temperature, followed by platinum sputtering of the dried samples with 30 mA sputter current at a 2.30 tooling factor for 40 s. The size and morphology of sputtered NPs were visualized at 5 kV using a field-emission scanning electron microscope (FE-SEM Hitachi/SU8010, Japan).
4.4. Murine Breast Cancer Cell Culture
Murine breast cancer-derived 4T1 cells were cultured in complete DMEM (cDMEM) medium (pH 7.4) containing 1% penicillin and streptomycin antibiotic and 10% FBS in a 25-cm2 flask, and the cell-containing flask was placed in a humidified incubator at 37 °C with 5% CO2. The cells were collected from the exponential growth phase and exposed to the trypsinization process, followed by repeated washing through centrifugation steps; they were then stained with trypan blue, counted using a hemocytometer, and then re-suspended in cDMEM medium at a concentration of 106 cells/mL. From this cell stock medium, the calculated volume of cell-containing medium was mixed with 100 µL of PBS (pH 7.4) to ensure 105 cells/100 µL for injecting in an animal model.
4.5. Tumor Regression Studies
Female Balb/c mice of 6–8 weeks old weighing 16–19 g were obtained from the School of Medicine and Health Science animal facility, Monash University. All animals were kept under 12-h/12-h light/dark conditions with free access to ab libitum and water. In vivo experiments were conducted in accordance with the approved animal ethics by the Monash Animal Ethics Committee (MARP/2016/126 and MUM/2018/12).
Approximately 1 × 10
5 4T1 cells/100 µL in PBS were subcutaneously injected on the mammary pad. Eleven days after inoculation, the mice were randomly divided into groups (
n = 5) when the tumor volume reached an average of 13.63 ± 2.68 mm
3. The length and width of an outgrowth tumor were estimated in mm by using a digital Vernier caliper over the period of 27 days, and the mice were sacrificed humanely via euthanasia, followed by measuring the weight of different organs of the mice. The body weights of mice with an induced tumor were measured in three-day intervals. The tumor volume data were presented as means ± SD of five different mice from each group. The following formula was used to calculate the tumor volume:
4.6. Fabrication of NPs and CYP-Loaded NPs for Tumor Model
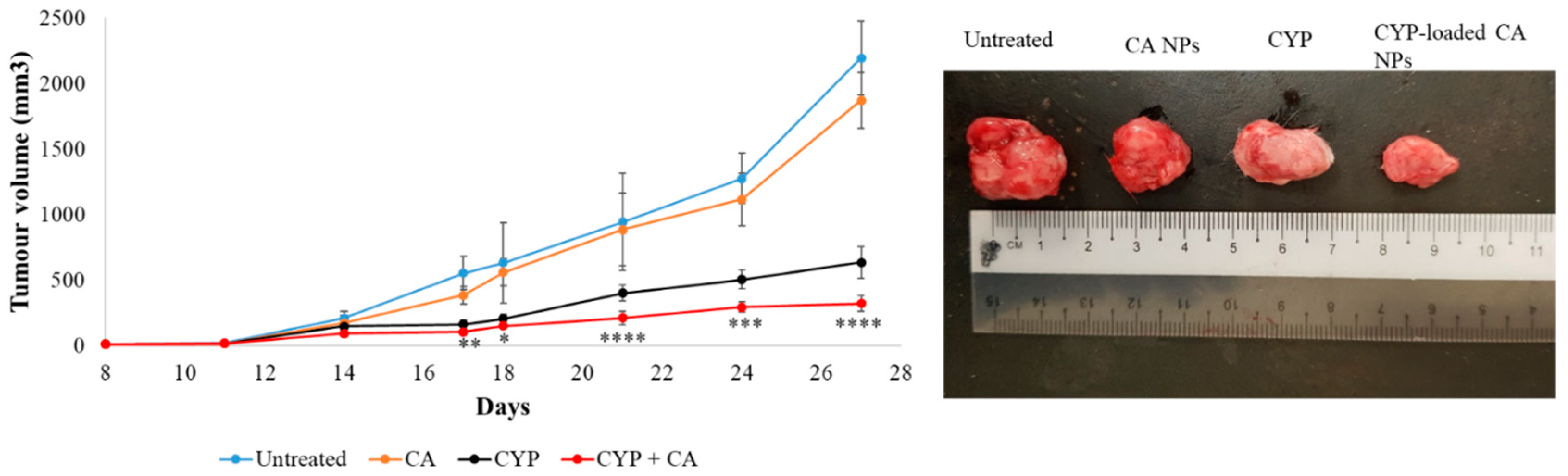
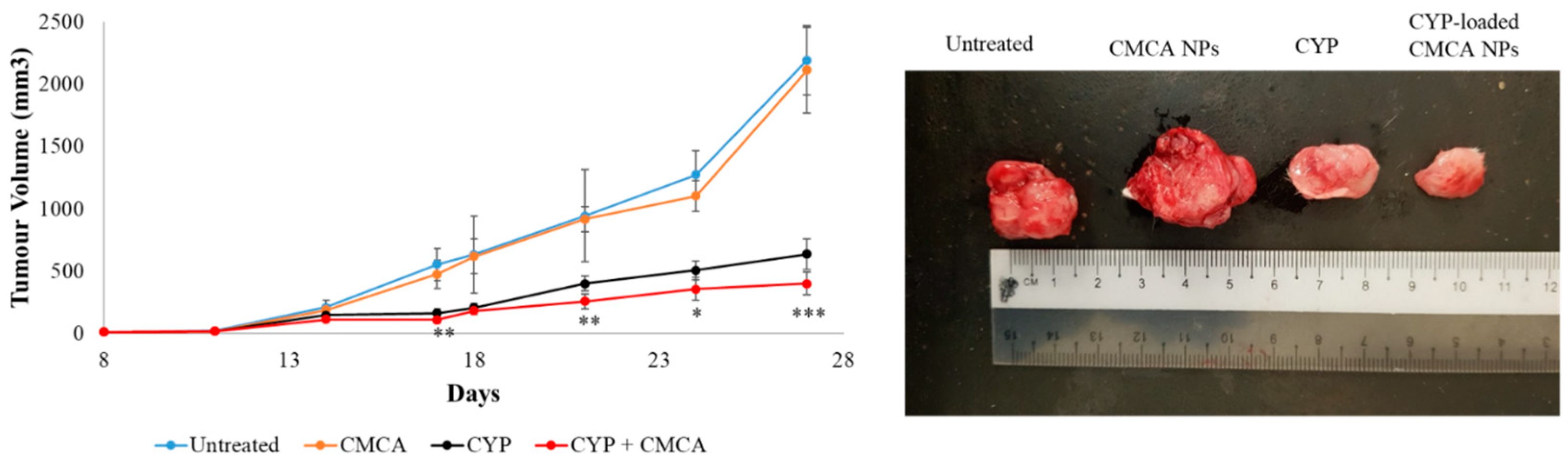
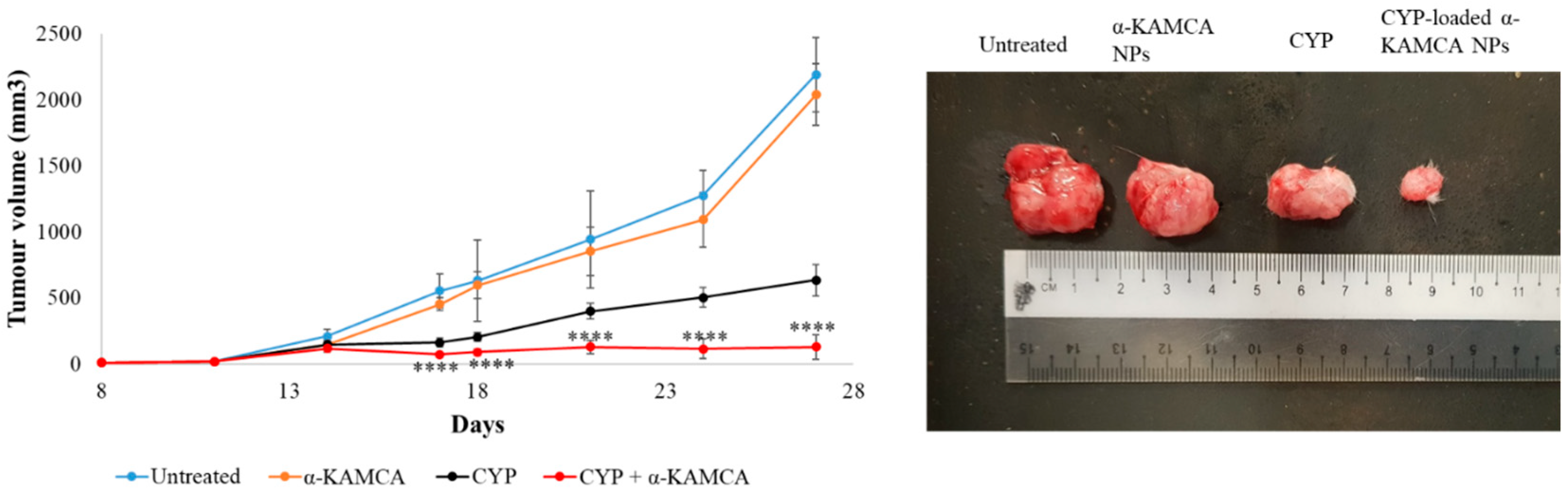
Filtered DMEM medium was prepared by following the abovementioned protocol. CA NPs were generated by adding 40 mM exogenous Ca2+ in 100 µL of filtered serum-free DMEM medium. Similarly, 40 mM exogenous Ca2+ and 10 mM sodium citrate were mixed in 100 µL of serum-free DMEM to formulate CMCA NPs. The α-KAMCA NPs were produced by combining 40 mM Ca2+ and 40 mM α-ketoglutaric acid in 100 µL of filtered serum-free DMEM solution. CYP at 198.79 µM (equivalent to 0.3 mg/kg BW) concentration was added to DMEM-containing PCR tubes along with the appropriate amount of exogenous Ca2+ in the presence or absence of citrate or α-ketoglutaric acid to synthesize CYP-loaded NPs. Also, CYP at 198.79 µM concentration was dissolved in serum-free filtered DMEM medium as a control. All the different mixtures with or without the drug were placed in an incubator for 30 min at 37 °C.
For the tumor regression study, one group of tumor-bearing mice was kept untreated as a negative control, the other tumor-bearing mice groups were treated with drug-free NPs or CYP only (dose of 0.3 mg/kg) as a positive control, and the remaining breast tumor-bearing mice were injected with CYP-loaded NPs (dose of 0.3 mg/kg) as a treatment group. The CYP solution, CYP-loaded CA NPs, CYP-loaded CMCA NPs, and CYP-loaded α-KAMCA NPs, as well as CA NPs, CMCA NPs, and α-KAMCA NPs, were injected intravenously using a 29G needle every two days with a total of three doses per mouse. The treatment was given at day 14 from tumor inoculation through the right or left caudal tail-vein, and the tumor volume was checked throughout the treatment period using a digital Vernier caliper.
4.7. Animal Biodistribution Studies
To investigate the tissue biodistribution of CA NPs, CMCA NPs, and α-KAMCA NPs, female Balb/c mice of 6–8 weeks old weighing 16–19 g were injected subcutaneously on the mammary pad with approximately 1 × 105 4T1 cells/100 µL in PBS. The mice were divided into groups (n = 5) when the tumor volume reached an average of 13.63 ± 2.68 mm3.
The DOX solution, DOX-loaded CA NPs, DOX-loaded CMCA NPs, and DOX-loaded α-KAMCA NPs were injected intravenously through the right or left caudal tail-vein with a DOX concentration of 5 mg/kg per dose. The mice were sacrificed after 2 h and 24 h of tail-vein injection (n = 5). The organs such as the liver, kidney, heart, spleen, lungs, and brain, as well as the tumor tissues, were isolated and kept in lysis buffer (pH 7.4) and kept at −150 °C in a freezer until further analysis. Additionally, the weights of all the organs and tumor tissues were measured for quantitative analysis of the presence of drug-loaded NPs.
Organs were homogenized and lysed with a lysis reagent (sodium fluoride and stable stock lysis buffer). The mixture was centrifuged at 8000 rpm at 4 °C for 50 min. The supernatant was collected in 1.5-mL microcentrifuge tubes. Then, 100 µL of supernatant was taken in black 96-well plates and subjected to fluorescence intensity measurement, quantified with an excitation wavelength of 485 nm and an emission wavelength of 535 nm using Perkin Elmer 2030 manager software attached to a 2030 multilabel reader victor ×5 (Perkin Elmer). The data were presented as means ± SD in relative fluorescence units (RFU)/mg of organ weight.
4.8. Blood Analysis
To determine the serum level of total drug-loaded NPs, blood samples of all the mice treated (n = 5) were collected by cardiac puncture using a 1-mL syringe with a 25G needle. A secondary method of euthanasia was performed after a cardiac puncture to sacrifice the animal. The blood samples were collected at two time points of analysis (2 h and 24 h of treatment). The blood samples were allowed to clot by leaving them at room temperature and then centrifuged at 10,000 rpm for 30 min at 4 °C to collect the supernatant serum. Afterward, 100 µL of each serum sample was collected and stored at −80 °C until it was assayed. The fluorescence emission of serum was measured using Perkin Elmer 2030 manager software attached to a 2030 multilabel reader victor X5 (Perkin Elmer) at 485/535 nm. The data were illustrated as means ± SD in RFU/100 µL of serum.
4.9. Toxicology Screening
According to the Organization for Economic Co-operation and Development (OECD) guidelines, the animals were screened for the toxicology study for the administration of different NPs [
74]. To analyze sub-chronic toxicity and to observe the mice’s overall health and well-being, CA NPs (40 mM exogenous Ca
2+ in 100 µL of filtered serum-free DMEM medium), CMCA NPs (40 mM exogenous Ca
2+ and 10 mM sodium citrate in 100 µL of serum-free DMEM), and α-KAMCA NPs (40 mM Ca
2+ and 40 mM α-ketoglutaric acid in in 100 µL of filtered serum-free DMEM solution) were administered intravenously through the right or left caudal tail-vein three times at two-day intervals, and the blood was collected by cardiac puncture after 27 days of the first treatment. For acute toxicity analysis, mice were treated intravenously with CA NPs (40 mM exogenous Ca
2+ in 100 µL of filtered serum-free DMEM media), CMCA NPs (40 mM exogenous Ca
2+ and 10 mM sodium citrate in 100 µL of serum-free DMEM), and α-KAMCA NPs (40 mM Ca
2+ and 40 mM α-ketoglutaric acid in 100 µL of filtered serum-free DMEM solution), and the blood sample was collected by cardiac puncture after 24 h of treatment.
During the sub-chronic study, the animals were well taken care throughout the 28 days, and any type of abnormality in behavior (e.g., less intake of water, less food intake, weight loss, less mobility, feces color change) of the treated mice was monitored. At each specific time point, the maximum volume of blood was extracted by cardiac puncture, and the mice were sacrificed humanely via euthanasia. The blood was collected into lithium heparin tubes (Vacutube, Vodice, Slovenia) and later transported in an ice box maintained at 4 °C to the Hematology and Biochemistry Clinical Laboratory, Faculty of Veterinary, University Putra Malaysia.
The test included the assessment of biomarkers for a blood electrolyte panel (Na+, K+, Cl−, Ca2+, PO43−), toxicity in the liver (i.e., alanine transaminase (ALT), alkaline phosphatase (ALP), total bilirubin (TBil), total protein (TP), and albumin (ALB) levels), kidney (i.e., urea level), and pancreas (i.e., amylase (Amy) level). All values and findings were compared between treated and untreated groups.
4.10. Protein Corona Analysis Using Liquid Chromatography and Mass Spectrometry (LC/MS)
CA NPs were prepared by adding 4 mM exogenous Ca2+ with incubation at 37 °C for 30 min. Similarly, CMCA and α-KAMCA NPs were synthesized by adding 1 mM sodium citrate and 4 mM α-ketoglutarate, respectively, along with 4 mM exogenous Ca2+, prior to incubation at 37 °C for 30 min. Then, 10% FBS was added to the particle suspensions before incubation at 37 °C for 10 min. The samples were then centrifuged at 13,000 rpm and 20 °C for 10 min. The supernatant was then removed and replaced with Milli Q water without mixing with the precipitate, and it was subjected to centrifugation under the same setting. The precipitate was collected and resuspended in 100 mL of 50 mM EDTA.
A C18 spin column (Thermo Fisher Scientific, USA) was used to perform liquid chromatography. The column was centrifuged at 1500× g relative centrifugal force (rcf) for 1 min, followed by discarding flow-through and repetition of a similar process. Then, 200 µL of equilibration solution (0.5% TFA in 5% ACN) was added and centrifuged at 1500× g rcf for 1 min. The NP samples were mixed with 1/3 sample buffer (2% TFA in 20% ACN) and loaded on top of the resin bed. The spin column was placed into a receiver and again centrifuged at 1500× g rcf for 1 min. Afterward, 200 µL of wash solution was run through the column, followed by the addition of 20 µL of elution buffer (70% ACN). After centrifugation, the collected flow-through (40 L for each sample) was dried in a Labogene vacuum evaporator (Bjarkesvej, Lillerød, Denmark) at 1000 rpm for 3 h at 50 °C.
4.11. In-Solution Digestion
The pellet formed from the evaporated sample was resuspended in 100 µL. Then, 50 μL was drawn out for further experimentation. It was added to 25 μL of ammonium bicarbonate, 25 µL of TFE, and 1 µL of DTT stock. The sample was then incubated at 60 °C for 60 min, and 4 µL of IAM was added; the sample was kept at room temperature in dark conditions for 60 min. The samples were mixed with 300 µL of water, 100 µL of ammonium bicarbonate, and 5 µL of trypsin, and they were incubated at 37 °C overnight. The prepared samples were subjected to mass spectroscopy Agilent MassHunter (Agilent Technologies), and the results were analyzed by protein identification through automated de novo sequencing using PEAKS Studio 7.0 (Bioinformatics Solution Inc.).
4.12. Dissolution Study of CA, CMCA, and α-KAMCA NPs at Different pH
CA NPs were generated using 20 mM Ca2+, CMCA NPs were prepared with 20 mM Ca2+ and 5 mM sodium citrate, and α-KAMCA NPs were synthesized using 20 mM Ca2+ and 20 mM alpha-ketoglutarate in 200-µL PCR tubes. All the salt mixtures in different PCR tubes were incubated at 37 °C for 30 min to formulate the corresponding NPs. After this, all the formulated NPs were suspended into 800 µL of DMEM medium at different pH (7.5, 7.0, 6.5, 6.0, 5.5, and 5.0), prior to the measurement of absorbance at 320 nm using an ultraviolet–visible light (UV–Vis) spectrophotometer. Data were represented as means ± SD of triplicates.
4.13. Statistical Analysis
Statistical significance was analyzed in drug-treated mice group versus drug-loaded NP-treated groups by one-way ANOVA (analysis of variance), followed by post hoc analyses using a Tukey multiple comparisons test with SPSS v23 for Windows. The minimal level of statistical significance was p < 0.05 with a 95% confidence interval (CI). The experimental data were presented as means ± SD (n = 5).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}