Molecular Determinants of Cancer Therapy Resistance to HDAC Inhibitor-Induced Autophagy



Abstract
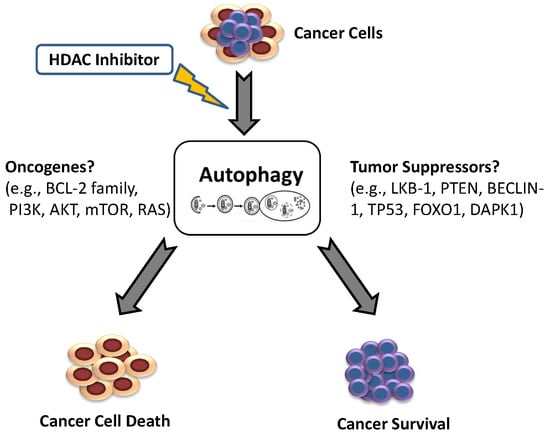
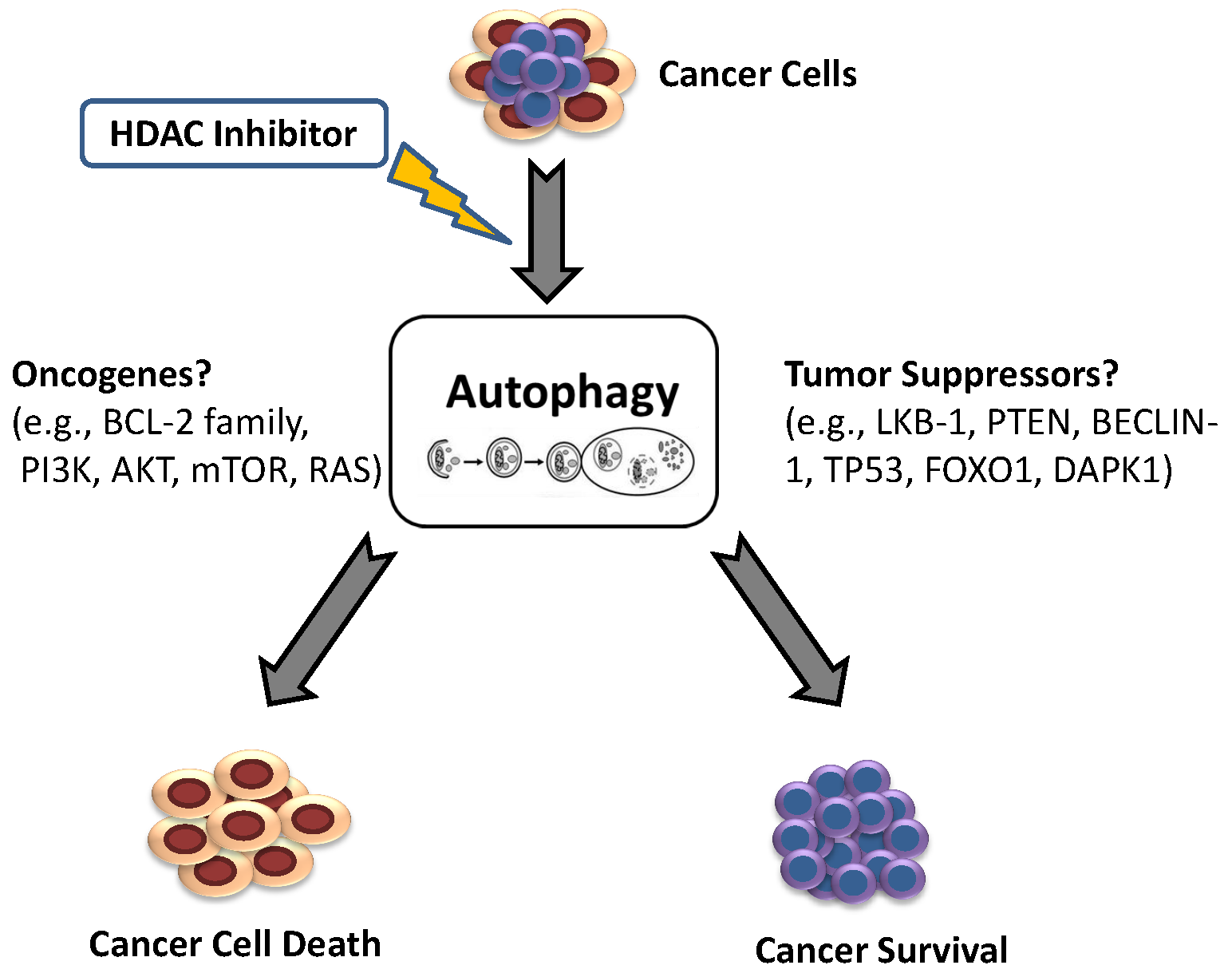
1. Introduction: Autophagy
2. The Oncogenic Role of Autophagy
3. Autophagy as a Therapeutic Target in Drug Resistance
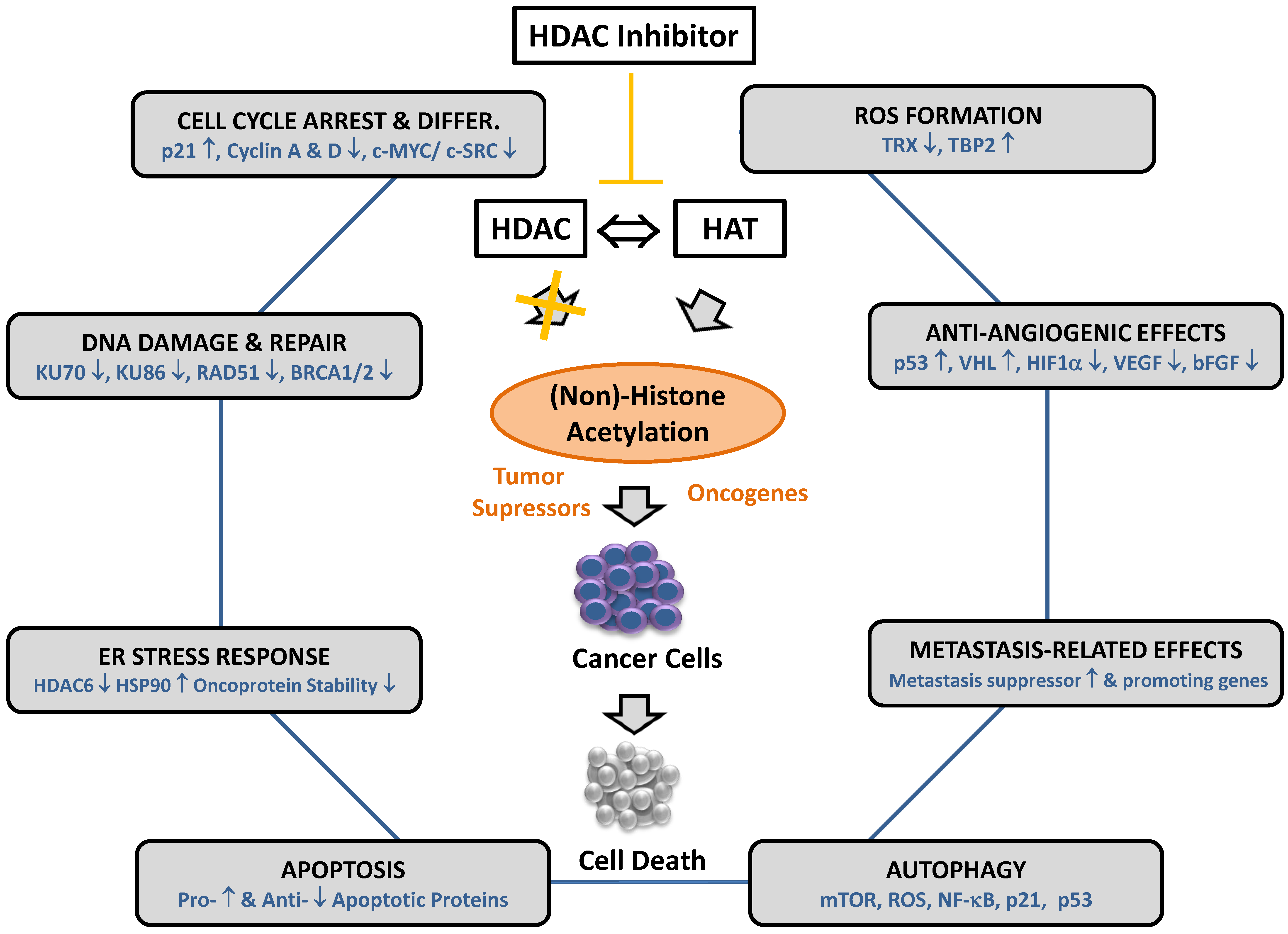
4. Histone Deacetylase Inhibitors and Their Effector Mechanisms
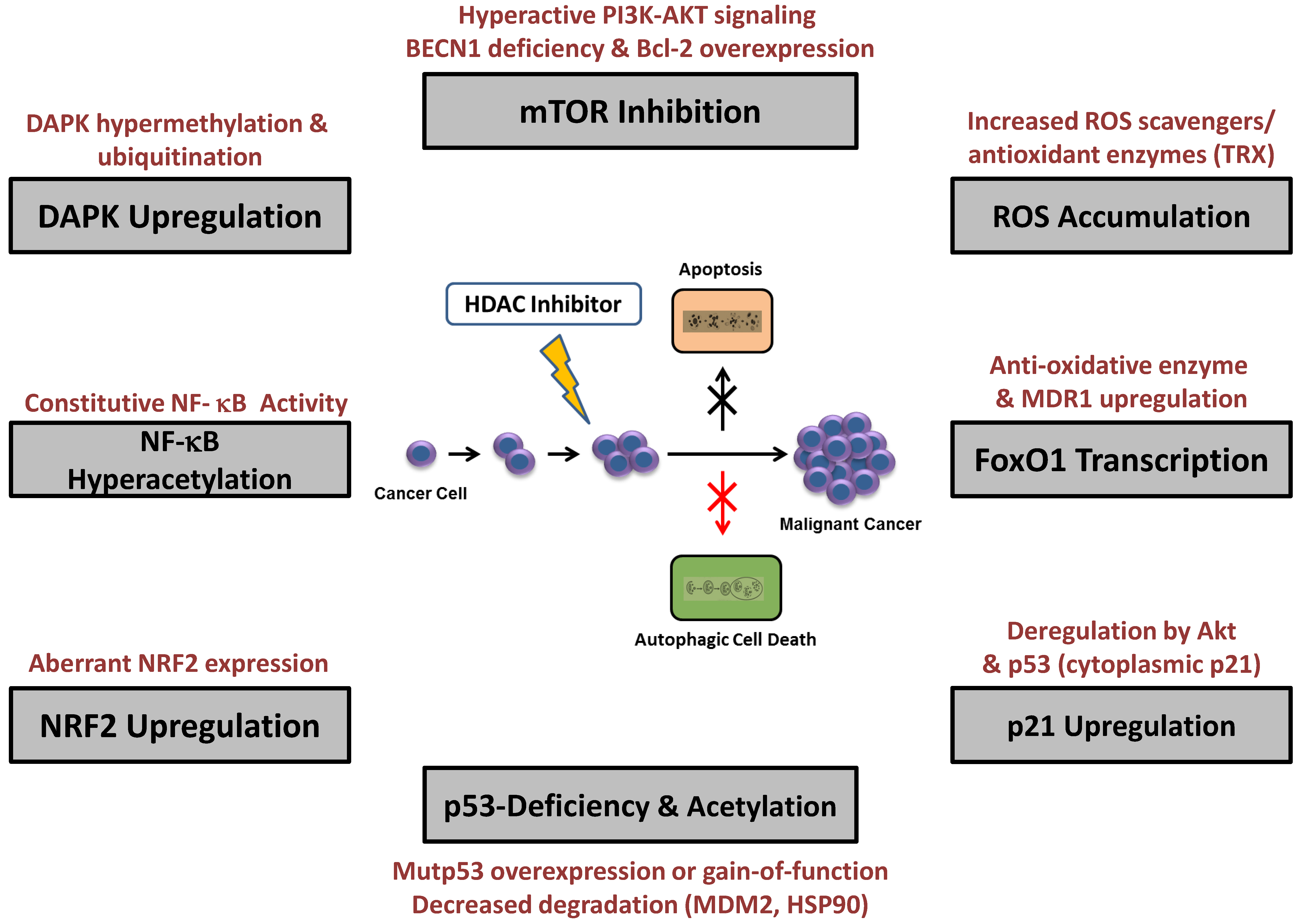
5. Resistance to HDAC Inhibitor-Induced Autophagy
5.1. mTOR Modulation
5.2. ROS Accumulation and Enhanced Antioxidant Expression
5.3. p21CIP/WAF-1 Upregulation
5.4. p53 Deficiency and Acetylation
5.5. NF-κB Hyperacetylation
5.6. FOXO1 Transcription
5.7. DAPK Upregulation
5.8. NRF2 Upregulation
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Dall, K.; Færgemann, N. Metabolic regulation of lifespan from a C. elegans perspective. Genes Nutr. 2019, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Kroemer, G. Autophagy in stress and disease. Cell Death Differ. 2015, 22, 365–366. [Google Scholar] [CrossRef][Green Version]
- Li, W.W.; Li, J.; Bao, J. Microautophagy: lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 305–309. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.; Morano, K.; Scott, S.; Klionsky, D. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1995, 131, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: a conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef]
- Reggiori, F.; Ungermann, C. Autophagosome maturation and fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Manuel, J.; Pedro, B.-S.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Yang, K.; Sathiyaseelan, P.; Ho, C.; Gorski, S. Evolution of tools and methods for monitoring autophagic flux in mammalian cells. Biochem. Soc. Trans. 2018, 46, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.; Walker, S.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Yanxiang Guo, J.; White, E. Autophagy, Metabolism, and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 73–78. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Eskelinen, E. The dual role of autophagy in cancer. Curr. Opin. Pharmacol. 2011, 11, 294–300. [Google Scholar] [CrossRef]
- Kubisch, J.; Türei, D.; Földvári-Nagy, L.; Dunai, Z.A.; Zsákai, L.; Varga, M.; Vellai, T.; Csermely, P.; Korcsmáros, T. Complex regulation of autophagy in cancer-integrated approaches to discover the networks that hold a double-edged sword. Semin. Cancer Biol. 2013, 23, 252–261. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A.; Foundation, A.; Brain, P.; States, U. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: a double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Lorin, S.; Hamaï, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta Mol. Cell Res. 2009. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Hunag, S.; Sinicrope, F.A. The role of autophagy in cancer: therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef]
- Morselli, E.; Galluzi, L.; Kepp, O.; Marino, G.; Michaud, M.; Vitale, I.; Maiuri, M.C.; Kroemer, G. Oncosuppressive functions of autophagy. Antioxdants Redox Signal. 2011, 14, 2251–2269. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget 2015, 6, 8474–8490. [Google Scholar] [CrossRef]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol. Cell. Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.L.M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef]
- Lopez, G.; Torres, K.; Lev, D. Autophagy blockade enhances HDAC inhibitors’ pro-apoptotic effects: Potential implications for the treatment of a therapeutic-resistant malignancy. Autophagy 2011, 7, 40–41. [Google Scholar] [CrossRef]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.-M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 68. [Google Scholar] [CrossRef]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Lahiri, P.; Zatloukal, K. Epigenetic silencing of apoptosis-inducing gene expression can be efficiently overcome by combined SAHA and TRAIL treatment in uterine sarcoma cells. PLoS ONE 2014, 9, e91558. [Google Scholar] [CrossRef]
- White, E. The role of autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Duprez, L.; Wirawan, E.; Vanden Berghe, T.; Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect. 2009, 11, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, L.; Zheng, H.; Mao, C.; Hu, W.; Xiong, K.; Wang, F.; Liu, C. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef]
- Maycotte, P.; Aryal, S.; Cummings, C.; Thorburn, J.; Morgan, M.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef]
- Li, L.; Xie, W.; Pan, D.; Chen, H.; Zhang, L. Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity of gastric cancer cells. Tumour Biol. 2016, 37, 653–659. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–323. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Autophagy as a target for cancer therapy: new developments. Cancer Manag. Res. 2012, 4, 357–365. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T.; Cleveland, J.L. Modulating autophagy for therapeutic benefit. Autophagy 2007, 3, 464–467. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelmann, A.; Debnath, J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, M.L.; Engedal, N.; Bøe, S.; Hokland, P.; Simonsen, A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8; 21) AML cells. Blood 2018, 122, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, K. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med. Chem. 2015, 7, 1535–1542. [Google Scholar] [CrossRef]
- Gammoh, N.; Marks, P.A.; Jiang, X. Curbing autophagy and histone deacetylases to kill cancer cells. Autophagy 2012, 8, 1521–1522. [Google Scholar] [CrossRef]
- Ying-Jie, L.; Yu-He, L.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Qadir, M.; Kwok, B.; Dragowska, W.; To, K.; Le, D.; Bally, M.; Gorski, S. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res. Treat. 2008, 112, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Samaddar, J.; Gaddy, V.; Duplantier, J.; Thandavan, S.; Shah, M.; Smith, M.; Browning, D.; Rawson, J.; Smith, S.; Barrett, J.; et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther. 2008, 7, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Yang, J.; Kung, H.; Shi, X.; Tilki, D.; Lara, P.J.; DeVere Whie, R.; Gao, A.; Evans, C. Targeting autphagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 2014, 33, 4521–4530. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Roy, S.; Lazar, A.; Wang, W.; McAuliffe, J.; Reynoso, D.; McMahon, J.; Taguchi, T.; Floris, G.; Debiec-Rychter, M.; et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc. Natl. Acad. Sci. USA 2010, 107, 14333–14338. [Google Scholar] [CrossRef]
- Amaravadi, R.; Lippincott-Schwartz, J.; Yin, X.; Weiss, W.; Takebe, N.; Timmer, W.; DiPaola, R.; Lotze, M.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; Ryan, K. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef]
- Liao, R.; Lin, Y.; Zhu, L. Molecular pathways involved in microRNA-mediated regulation of multidrug resistance. Mol. Biol. Rep. 2018, 45, 2913–2923. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Chrun, E.; Modolo, F.; Ivan, F.D. Histone modifications: A review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol. Pract. 2017, 213, 1329–1339. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef]
- Vrana, J.; Decker, R.; Johnson, C.; Wang, Z.; Jarvis, W.; Richon, V.; Ehinger, M.; Fisher, P.; Grant, S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18, 7016–7025. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Douglas, L.; Cleveland, J.L.; Humphreys, R.; Houghton, J.A. Histone deacetylase inhibitors enhance lexatumumab-induced apoptosis via a p21 Cip1 -dependent decrease in survivin levels. Cancer Res. 2007, 67, 6987–6995. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Yu, C.; Reese, E.; Ahmed, W.; Hirsch, K.; Dent, P.; Grant, S. Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of p21 CIP1/WAF1 induction rather than AKT inhibition. Oncogene 2003, 22, 6231–6242. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.J.; Pavey, S.; Warrener, R.; Hunter, L.K.; Piva, T.J.; Musgrove, E.A.; Saunders, N.; Parsons, P.G.; Gabrielli, B.G. Up-regulation of p21 WAF1/CIP1 by histone deacetylase inhibitors reduces their cytotoxicity. Mol. Pharmacol. 2001, 60, 828–837. [Google Scholar] [PubMed]
- Richon, V.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, B.; Rifkind, R.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21 WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 1–6. [Google Scholar] [CrossRef]
- Souleimani, A.; Asselin, C. Regulation of c-myc expression by sodium butyrate in the colon carcinoma cell line Caco-2. FEBS Lett. 1993, 326, 45–50. [Google Scholar] [CrossRef]
- Hirsch, C.L.; Smith-Windsor, E.L.; Bonham, K. Src family kinase members have a common response to histone deacetylase inhibitors in human colon cancer cells. Int. J. Cancer 2006, 118, 547–554. [Google Scholar] [CrossRef]
- Qiu, L.; Burgess, A.; Fairlie, D.; Leonard, H.; Parsons, P.; Gabrielli, B. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2001, 98, 10833–10838. [Google Scholar] [CrossRef]
- Lee, J.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Warrener, R.; Beamish, H.; Burgess, A.; Waterhouse, N.J.; Giles, N.; Fairlie, D.; Gabrielli, B. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB J. 2003, 17, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Zhou, X.; Xu, W.-S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21 CIP1/WAF1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef]
- Lin, K.; Wang, Y.; Chen, C.; Ho, C.; Su, W.; Jou, Y. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef]
- Pulukuri, S.; Gorantla, B.; Rao, J. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator. J. Biol. Chem. 2007, 282, 35594–35603. [Google Scholar] [CrossRef]
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs mediate HIF-1a stability through PHD2-dependent mechanism, while HDAC6, a class IIb member, promotes HIF1a transcriptional activity in nucleus pulposus cells of the intervertebral disc. J. Bone Miner. Res. 2016, 31, 1287–1299. [Google Scholar] [CrossRef]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.-E.; Lee, S.-W.; Moon, E.-J.; Kim, H.-S.; Lee, S.-K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef]
- Liu, T.; Kuljaca, S.; Tee, A.; Marshall, G.M. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat. Rev. 2006, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Mudduluru, G.; Antony, S.; Vashistha, S.; Ajitkumar, P.; Somasundaram, K. Expression profiling of sodium butyrate (nab)-treated cells: Identification of regulation of genes related to cytokine signaling and cancer metastasis by nab. Oncogene 2004, 23, 6304–6315. [Google Scholar] [CrossRef] [PubMed]
- McGarry, L.; Winnie, J.; Ozanne, B. Invasion of v-fos(fbr)-transformed cells is dependent upon histone deacetylase activity and suppression of histone deacetylase regulated genes. Oncogene 2004, 23, 5284–5392. [Google Scholar] [CrossRef]
- Kim, S.; Ahn, S.; Han, J.; Lee, H.; Lee, H.; Lee, Y.; Kim, M.; Kim, K.; Kim, W.; Hong, S. Apicidin is a histone deacetylase inhibitor with anti-invasive and anti-angiogenic potentials. Biochem. Biophys. Res. Commun. 2004, 315, 964–970. [Google Scholar] [CrossRef]
- Munshi, A.; Kurland, J.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef]
- Subramanian, C.; Opipari, A.W.; Bian, X.; Castle, V.P.; Kwok, R.P. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 4842–4847. [Google Scholar] [CrossRef]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Fröhlich, L.F. p53-mediated molecular control of autophagy in tumor cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Fröhlich, L.F. Epigenetic targeting of autophagy via HDAC inhibition in tumor cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone deacetylase inhibitor-induced autophagy in tumor cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Fröhlich, L. Regulation of HDACi-triggered autophagy by the tumor suppressor protein p53. In Genes and Cancer; Lemamy, G.-J., Ed.; InTech Open: Rijeka, Croatia, 2019; pp. 1–26. ISBN 978-1-78984-427-6. [Google Scholar]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC family members intertwined in the regulation of autophagy: A druggable vulnerability in aggressive tumor entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [Google Scholar] [CrossRef]
- Rikiishi, H. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. J. Biomed. Biotechnol. 2011, 2011, 830260. [Google Scholar] [CrossRef]
- Di Giacomo, V.; Di Valerio, V.; Rapino, M.; Bosco, D.; Cacciatore, I.; Ciulla, M.; Marrazzo, A.; Fiorito, J.; Di Stefano, A.; Cataldi, A. MRJF4, a novel histone deacetylase inhibitor, induces p21 mediated autophagy in PC3 prostate cancer cells. Cell. Mol. Biol. 2015, 61, 17–23. [Google Scholar]
- Yamamoto, S.; Tanaka, K.; Sakimura, R.; Okada, T.; Nakamura, T.; Li, Y.; Takasaki, M.; Nakabeppu, Y.; Iwamoto, Y. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 2008, 28, 1585–1591. [Google Scholar]
- Saveria, M.; Montani, G.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; Orazi, G.D.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Wang, J.; Kim, T.H.; Ahn, M.Y.; Lee, J.; Jung, J.H.; Choi, W.S.; Lee, B.M.; Yoon, K.S.; Yoon, S.; Kim, H.S. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 1101–1109. [Google Scholar] [CrossRef]
- Park, E.Y.; Woo, Y.; Kim, S.J.; Kim, D.H.; Lee, E.K.; De, U.; Kim, K.S.; Lee, J.; Jung, J.H.; Ha, K.T.; et al. Anticancer effects of a new SIRT inhibitor, MHY2256, against human breast cancer MCF-7 cells via regulation of MDM2-p53 binding. Int. J. Biol. Sci. 2016, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- De, U.; Son, J.Y.; Sachan, R.; Park, Y.J.; Kang, D.; Yoon, K.; Lee, B.M.; Kim, I.S.; Moon, H.R.; Kim, H.S. A new synthetic histone deacetylase inhibitor, MHY2256, induces apoptosis and autophagy cell death in endometrial cancer cells via p53 acetylation. Int. J. Mol. Sci. 2018, 19, 2743. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Ahn, J.W.; Kim, H.S.; Lee, J.; Yoon, J.H. Apicidin inhibits cell growth by downregulating IGF-1R in salivary mucoepidermoid carcinoma cells. Oncol. Rep. 2015, 33, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef]
- Chiao, M.-T.; Cheng, W.-Y.; Yang, Y.-C.; Shen, C.-C.; Ko, J.-L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef]
- Liu, Y.L.; Yang, P.; Shun, C.; Wu, M.; Weng, J.; Chen, C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef]
- Hrzenjak, A.; Moinfar, F.; Kremser, M.-L.; Strohmeier, B.; Petru, E.; Zatloukal, K.; Denk, H. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol. Cancer 2010, 9, 49. [Google Scholar] [CrossRef]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; Van Grevenynghe, J.; Belgnaoui, S.M.; Nguyen, T.L.A.; Di Lenardo, T.; Semmes, O.J.; Lin, R.T.; et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-kappa B-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef]
- Long, J.; Zhao, J.; Yan, Z.; Liu, Z.; Wang, N. Antitumor effects of a novel sulfur-containing hydroxamate histone deacetylase inhibitor H40. Int. J. Cancer 2009, 124, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Adachi, S.; Matsubara, H.; Imai, T.; Yui, Y.; Mizushima, Y. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer Res. 2009, 67, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.; Yang, N.; Lin, Q.; Xia, D.; Shen, H. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yi, Y.; Xia, G.; Zhao, Y.; Yu, Y.; Li, L.; Hua, C.; He, B.; Yang, B.; Yu, C.; et al. Nrf2-miR-129-3p-mTOR Axis Controls an miRNA Regulatory Network Involved in HDACi-Induced Autophagy. Mol. Ther. 2019, 27, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Gandesiri, M.; Chakilam, S.; Ivanovska, J.; Benderska, N.; Ocker, M.; Di Fazio, P.; Feoktistova, M.; Gali-Muhtasib, H.; Rave-Fränk, M.; Prante, O.; et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2012, 17, 1300–1315. [Google Scholar] [CrossRef]
- Yi, C.; Ma, M.; Ran, L.; Zheng, J.; Tong, J.; Zhu, J.; Ma, C.; Sun, Y.; Zhang, S.; Feng, W.; et al. Function and molecular mechanism of acetylation in autophagy regulation. Science 2012, 336, 474–477. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.C.; Roland, O.; Cherrier-De Wilde, S.; Plawny, L.; Van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef]
- El-Khoury, V.; Moussay, E.; Janji, B.; Palissot, V.; Aouali, N.; Brons, N.H.; Van Moer, K.; Pierson, S.; Van Dyck, E.; Berchem, G. The histone deacetylase inhibitor MGCD0103 induces apoptosis in B-cell chronic lymphocytic leukemia cells through a mitochondria-mediated caspase activation cascade. Mol. Cancer Ther. 2010, 9, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Hrzenjak, A.; Kremser, M.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, C.; Xue, R.; Hsueh, W.; Pan, P.; Chen, Z.; Wang, S.; McNutt, M.; Gu, J. Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int. J. Biochem. Cell Biol. 2008, 40, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Podsypanina, K.; Lee, R.; Politis, C.; Hennessey, I.; Crance, A.; Puc, J.; Neshat, M.; Wang, H.; Yang, L.; Gibbons, J.; et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/- mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10320–10325. [Google Scholar] [CrossRef] [PubMed]
- Neshat, M.; Mellinghoff, I.; Tran, C.; Stiles, B.; Thomas, G.; Petersen, R.; Frost, P.; Gibbons, J.; Wu, H.; Sawyers, C. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. USA 2001, 98, 10314–10319. [Google Scholar] [CrossRef]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef]
- Ahn, M.-Y.; Ahn, S.-G.; Yoon, J.-H. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncol. 2011, 47, 1032–1038. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Reed, J.C.; Cuddy, M.; Slabiak, T.; Croce, C.M.; Nowell, P.C. Oncogenic potential of bcl-2 demonstrated by gene transfer. Nature 1988, 336, 259–261. [Google Scholar] [CrossRef]
- Reed, J.C.; Kitada, S.; Takayama, S.; Miyashita, T. Regulation of chemoresistance by the bcl-2 oncoprotein in non-Hodgkin’s lymphoma and lymphocytic leukemia cell lines. Ann. Oncol. 1994, 5, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Kitada, S.; Takayama, S.; De Riel, K.; Tanaka, S.; Reed, J.C. Reversal of chemoresistance of lymphoma cells by antisense-mediated reduction of bcl-2 gene expression. Antisense Res. Dev. 1994, 4, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R.J.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Yeung, P.L.; Chiang, A.K.S. Induction of MAPK- and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget 2015, 7, 1–14. [Google Scholar] [CrossRef]
- Zhan, Y.; Gong, K.; Chen, C.; Wang, H.; Li, W. P38 MAP kinase functions as a switch in MS-275-induced reactive oxygen species-dependent autophagy and apoptosis in human colon cancer cells. Free Radic. Biol. Med. 2012, 53, 532–543. [Google Scholar] [CrossRef]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Santoro, R.; Strano, S.; Blandino, G. Transcriptional regulation by mutant p53 and oncogenesis. Subcell. Biochem. 2014, 85, 91–103. [Google Scholar] [CrossRef]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4, 2996. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M. Mutant p53 in cancer: Accumulation, gain-of-function, and therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M. Are p27 and p21 cytoplasmic oncoproteins? Cell Cycle 2002, 1, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Abukhdeir, A.; Park, B. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Zhou, B.; Liu, X.; Heung, Y.; Chau, J.; Chu, E.; Li, S.; Jiang, C.; Un, F.; Yen, Y. Ribonucleotide reductase small subunit p53R2 facilitates p21 induction of G1 arrest under UV irradiation. Cancer Res. 2007, 67, 16–21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Compagno, M.; Lim, W.; Grunn, A.; Nandula, S.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kB in diffuse large B cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Cho, K.; Choi, H.; Han, H.; Kang, K. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis 2008, 29, 1837–1844. [Google Scholar] [CrossRef]
- Goto, T.; Takano, M.; Hirata, J.; Tsuda, H. The involvement of FOXO1 in cytotoxic stress and drug-resistance induced by paclitaxel in ovarian cancers. Br. J. Cancer 2008, 98, 1068–1075. [Google Scholar] [CrossRef]
- Ramanathan, B.; Jan, K.; Chen, C.; Hour, T.; Yu, H.; Pu, Y. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res. 2005, 65, 8455–8460. [Google Scholar] [CrossRef]
- Ogawa, T.; Liggett, T.; Melnikov, A.; Monitto, C.; Kusuke, D.; Shiga, K.; Kobayashi, T.; Horii, A.; Chatterjee, A.; Levenson, V.; et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle 2012, 11, 1656–1663. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Yuan, W.-C.; Ho, H.-C.; Chen, C.-H.; Shih, H.-M.; Chen, R.-H. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2010, 29, 1748–1761. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.; Motohashi, H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Ann. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Chakraborty, A.R.; Basseville, A.; Luchenko, V.; Zhan, Z.; Bates, S.E. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol. Pharmacol. 2012, 8, 2021–2031. [Google Scholar] [CrossRef]
- Fantin, V.; Richon, V. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res. 2007, 13, 7237–7242. [Google Scholar] [CrossRef]
- Lopez, G.; Torres, K.; Liu, J.; Hernandez, B.; Young, E.; Belousov, R.; Bolshakov, S.; Lazar, A.J.; Slopis, J.M.; McCutcheon, I.E.; et al. Autophagic survival in resistance to histone deacetylase inhibitors: novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011, 71, 185–196. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choy, M.L.; Marks, P.A. Mechanism of resistance to histone deacetylase inhibitors. Adv. Cancer Res. 2012, 116, 39–86. [Google Scholar] [CrossRef]
- Hippert, M.M.; Toole, P.S.O.; Thorburn, A. Autophagy in cancer: Good, bad, or both? Cancer Res. 2006, 66, 9349–9352. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Simonsen, A. Autophagy-Friend or foe in the treatment of fusion protein-associated leukemias? Autophagy 2013, 9, 2175–2177. [Google Scholar] [CrossRef][Green Version]
- Robey, R.; Zhan, Z.; Piekarz, R.; Kayastha, G.; Fojo, T.; Bates, S. Increased MDR1 expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176). Clin. Cancer Res. 2006, 12, 1547–1555. [Google Scholar] [CrossRef]
- Piekarz, R.; Robey, R.; Zhan, Z.; Kayastha, G.; Sayah, A.; Abdeldaim, A.; Torrico, S.; Bates, S. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood 2004, 103, 4636–4643. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Huang, Y.; Dai, Z.; Sadee, W.; Chen, J.; Liu, S.; Marcucci, G.; Byrd, J.; Covey, J.; Wright, J.; et al. Chemoresistance to depsipeptide FK228 is mediated by reversible MDR1 induction in human cancer cell lines. J. Pharmacol. Exp. Ther. 2005, 314, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Epping, M.; Meijer, L.; Bos, J.; Bernards, R. UNC45A confers resistance to histone deacetylase inhibitors and retinoic acid. Mol. Cancer Res. 2009, 7, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Mickley, L.; Bates, S.; Richert, N.; Currier, S.; Tanaka, S.; Foss, F.; Rosen, N.; Fojo, A. Modulation of the expression of a multidrug resistance gene (mdr-1/P-gylcoprotein) by differentiating agents. J. Biol. Chem. 1989, 264, 18301–18340. [Google Scholar]
- Frommel, T.; Coon, J.; Tsuruo, T.; Roninson, I. Variable effects of sodium butyrate on the expression and function of the mdr-1 (P-glycoprotein) gene in colon carcinoma cell lines. Int. J. Cancer 1993, 55, 297–302. [Google Scholar] [CrossRef]
- Fantin, V.; Loboda, A.; Paweletz, C.; Hendrickson, R.; Pierce, J.; Roth, J.; Li, L.; Gooden, F.; Korenchuck, S.; Hou, X.; et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008, 68, 3785–3794. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef]
- Noda, T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef]
- Saxton, R.; Sabatini, D. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D. mTOR signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011593. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Jacinto, E.; Wullschlager, S.; Lorberg, A.; Crespo, J.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Ma, X.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and elF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Luo, Z.; Saha, A.K.; Xiang, X.; Ruderman, N.B. AMPK, the metabolic syndrome and cancer. Trends Pharmacol. Sci. 2005, 26, 69–76. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Kun-Liang, G. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.; Kim, Y.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- Lin, S.; Li, T.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.; Lian, G.; Liu, Q.; Ruan, K.; et al. GSK3-TIP60-ULK1 signaling links growth factor deprivation to autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef]
- Aoki, M.; Blazek, E.; Vogt, P. A role for the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc. Natl. Acad. Sci. USA 2001, 98, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Meijer, A.J.; Codogno, P. Autophagy and p70S6 kinase. Autophagy 2005, 1, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Balakumaran, B.S.; Herbert, J.T.; Febbo, P.G. MYC activity mitigates response to rapamycin in prostate cancer through 4EBP1-mediated inhibition of autophagy. Autophagy 2010, 6, 281–282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef]
- Maiuri, M.; Le Toumelin, G.; Criollo, A.; Rain, J.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.; Mizushima, N.; Packer, M.; Schneider, M.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef]
- Yuan, T.; Cantley, L. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- Shaw, R.; Cantley, L. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Akar, U.; Chaves-Reyez, A.; Barria, M.; Tari, A.; Sanguino, A.; Kondo, Y.; Kondo, S.; Arun, B.; Lopez-Berestein, G.; Ozpolat, B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 2008, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Szumiel, I. Autophagy, reactive oxygen species and the fate of mammalian cells. Free Radic. Res. 2011, 45, 253–265. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Novak, I. Mitophagy: A complex mechanism of mitochondrial removal. Antioxidants Redox Signal. 2012, 17, 794–802. [Google Scholar] [CrossRef]
- Feng, D.; Liu, L.; Zhu, Y.; Chen, Q. Molecular signaling toward mitophagy and its physiological significance. Exp. Cell Res. 2013, 319, 1697–1705. [Google Scholar] [CrossRef]
- Ranganathan, A.; Zhang, L.; Adam, A.; Aquirre-Ghiso, J. Functional coupling of p38-induced upregulation of BiP and activation of RNA-dependent protein kinase-like endoplasmatic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef]
- Warr, M.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegue, E. FOXO3 directs a protective autophagy program in hematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef]
- Rouschop, K.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Alexander, A.; Cai, S.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.; Inoki, K.; Guan, K.; Shen, J.; Person, M.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuck, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the crossroads between different types of HDAC-inhibitor-mediated cancer cell death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagoskonny, M.V.; Bates, S.E. P21-dependent G1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Wang, X.; Zhenchuk, A.; Wimann, K.; Albertioni, F. Regulation of p53R2 and its role as potential target for cancer therapy. Cancer Lett. 2009, 276, 1–7. [Google Scholar] [CrossRef]
- Schepers, H.; Geugien, M.; Eggen, B.; Vellenga, E. Constitutive cytoplasmic localization of p21Waf1/Cip1 affects the apoptotic process in monocytic leukaemia. Leukemia 2003, 17, 2113–2121. [Google Scholar] [CrossRef]
- Crighton, D.; O’Prey, J.; Bell, H.; Ryan, K. p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ. 2007, 14, 1071–1079. [Google Scholar] [CrossRef]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–10. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.-G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Guy, A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2011, 327, 1223–1228. [Google Scholar] [CrossRef]
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 2013, 62, 70–77. [Google Scholar] [CrossRef]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2010, 16, 1135–1145. [Google Scholar] [CrossRef]
- Pimkina, J.; Humbey, O.; Zilfou, J.; Jarnik, M.; Murphy, M. ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. P53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. J. Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.; Galluzi, L.; Morselli, E.; Kepp, O.; Malik, S.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181185. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53´s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef]
- Tasdemir, E.M.; Maiuri, C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Segerba, D.; Bergman, J.; Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; So, M.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.; Issaeva, N.; Selivanova, G.; Wiman, K.G. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the National Cancer Institute database. Carcinogenesis 2002, 23, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.; Coffey, H.; Morin, M.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed]
- Floquet, C.; Deforges, J.; Rousset, J.-P.; Bidou, L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011, 39, 3350–3362. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.; Moll, U. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Diff. 2011, 18, 1904–1913. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Trostel, S.; Kayastha, G.; Demidenko, Z.N.; Vassilev, L.T.; Romanova, L.Y.; Bates, S.; Fojo, T. Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res. 2005, 65, 7386–7392. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Schulz, R. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef]
- Garufi, A.; Pucci, D.; Orazi, V.D.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; Orazi, G.D. Degradation of mutant p53H175 protein by Zn (II) through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.; Davis, R.; Pierce, J.; Hepperle, M.; Xu, Y.; Hottelet, M.; Nong, Y.; Wen, D.; Adams, J.; Dang, L.; et al. Small molecule inhibitors of IkB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin. Cancer Res. 2005, 11, 28–40. [Google Scholar] [PubMed]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive NF-?B activation in B-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Senovilla, L.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; Tailler, M.; Delahaye, N.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar] [CrossRef]
- Mortezaee, K.; Najafi, M.; Farhood, B.; Ahmadi, A.; Shabeeb, D.; Musa, A.E. NF-κB targeting for overcoming tumor resistance and normal tissues toxicity. J. Cell. Physiol. 2019, 234, 17187–17204. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B. FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Kops, G.; Dansen, T.; Polderman, P.; Saarloos, I.; Wirtz, K.; Coffer, P.; Huang, T.; Bos, J.; Medema, R.; Burgering, B. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Dijkers, P.; Medema, R.; Pals, C.; Banerji, L.; Thomas, N.; Lam, E.; Burgering, B.M.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 2000, 20, 9138–9148. [Google Scholar] [CrossRef]
- Dijkers, P.; Medema, R.; Lammers, J.; Koenderman, L.; Coffer, P. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 2000, 10, 1201–1204. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Bommareddy, A.; Kim, S.; Sehrawat, A.; Hahm, E.; Singh, S. Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS ONE 2012, 7, e32597. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pan, X.; Xu, Y.; Xiao, Y.; An, Y.; Tie, L.; Pan, Y.; Li, X. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012, 8, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Tindall, D. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.; Myatt, S.; Lam, E. Many forks in the path: cycling with FoxO. Oncogene 2008, 27, 2300–2311. [Google Scholar] [CrossRef]
- Lin, A.; Piao, H.; Zhuang, L.; Sarbassov, D.; Ma, L.; Gan, B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacological inhibition of the PI3K-AKT pathway. Cancer Res. 2014, 74, 1682–1693. [Google Scholar] [CrossRef]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: structure, function and beyond. Annu. Rev. Biochem 2006, 75, 189–210. [Google Scholar] [CrossRef]
- Deiss, L.; Feinstein, E.; Berissi, H.; Cohen, O.; Kimchi, A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995, 9, 15–30. [Google Scholar] [CrossRef]
- Levin-Salomon, V.; Bialik, S.; Kimchi, A. DAP-kinase and autophagy. Apoptosis 2014, 19, 346–356. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Kimchi, A. DAP-kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ. 2007, 14, 1908–1915. [Google Scholar] [CrossRef]
- Wu, J.; Hu, C.; Gu, Q.; Li, Y.; Song, M. Trichostatin A sensitizes cisplatin-resistant A549 cells to apoptosis by up-regulating death-associated protein kinase. Acta Pharmacol. Sin. 2010, 31, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurakawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, P.; Schmidt, V.; Smole, C.; Kufferath, I.; Denk, H.; Strnad, P.; Rülicke, T.; Fröhlich, L.F.; Zatloukal, K. P62/Sequestosome-1 Is indispensable for maturation and stabilization of Mallory-Denk bodies. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef]
- Yang, H.; Ni, H.-M.; Guo, F.; Ding, Y.; Shi, Y.-H.; Lahiri, P.; Fröhlich, L.F.; Rülicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/p62 protein is associated with autophagic removal of excess hepatic endoplasmic reticulum in mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef]
- Bao, L.J.; Jaramillo, M.C.; Zhang, Z.B.; Zheng, Y.X.; Yao, M.; Zhang, D.D.; Yi, X.F. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 1502–1513. [Google Scholar]
- Belleza, I.; Giambanco, I.; Minelli, A.; Doanto, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta. Mol. Cell. Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: An update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Wagner, J.M.; Hackanson, B.; Lübbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenetics 2010, 1, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.; Heaney, M.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical experience with intravenous and oral formulation of the hovel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006, 24, 166–172. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Autophagic Regulation | HDACi | Cancer Cell Type | Ref. | Mechanisms of Resistance | Ref. |
|---|---|---|---|---|---|
| mTOR Inhibition | SAHA | ESS-1 | [49,153] | Constitutive PI3K-AKT signaling -AKToverexpression -PI3K mutations -Loss of PTEN, ARHI, LKB-1 -other upstream factors Beclin-1 deficiency Bcl-2 overexpression | |
| SAHA | Glioblastoma | [136] | |||
| Butyrate, SAHA | HelaS3 | [135,154] | |||
| SAHA, OSU-HAD, C42 | HCC, Hep3B, HepG2 | [139] | [155,156,157,158] | ||
| SAHA | Jurkat T-cells | [137] | |||
| SAHA | Gliobastoma SC | [138] | |||
| Apicidin | Salivary MEC | [134,159] | [160,161,162,163] | ||
| * MGCD0103 | Primary CLL | [151,152] | |||
| * SAHA, TSA, VPA, MS-275, JQ2 | DS-AMKL cells | [64] | |||
| ROS Accumulation | SAHA | Jurkat T-cells | [137] | Increased levels of ROS scavengers/antioxidant enzymes (TRX) | [164,165] |
| SAHA | CMLL | [58] | |||
| FK228 | Gastric carcinoma | [165] | |||
| M-275 | HCT116 | [166] | |||
| VPA, SAHA | AML (Kasumi-1) | [62] | |||
| VPA, TSA | PaCa44, Panc1 | [130] | |||
| p53 Acetylation & Deficiency | Sirtinol | MCF-7 | [131] | Overexpression/GOF of mutant p53 Decreased degradation of p53 (MDM2, HSP90) | [167,168,169,170,171] |
| MHY2256 | MCF-7 | [132] | |||
| MHY2256 | Endometrial | [133] | |||
| SAHA | ESS-1 | [49] | |||
| VPA, TSA | PaCa44, Panc1 | [130] | |||
| p21 Upregulation | SAHA, H40 | PC-3M, HL-60 | [142] | Deregulated phosphorylation by Akt1 Deregulation by p53 (p53R2; cytoplasmic p21) | [172,173,174,175] |
| MRJF4 | PC3 | [128] | |||
| Apicidin | Salivary MEC | [134,159] | |||
| VPA, TSA | PaCa44, Panc1 | [130] | |||
| NF-κB Hyper-acetylation | SAHA, MS-275 | PC3 | [141] | (Constitutive) NF-κB upregulation | [176] |
| FOXO1 Transcription | LBH589 | HepG2, HCT116 | [145] | MDR1 upregulation Anti-oxidative enzyme upregulation | [177,178,179] |
| DAPK Upregulation | SAHA, TSA, LBH589, JQ2 | HCT116 | [147] | DAPK hypermethylation & ubiquitination | [180,181] |
| NRF2 upregulation | SAHA, TSA | Huh-7, MGC-803 | [146] | Aberrant NRF2 expression | [182,183,184] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mrakovcic, M.; Fröhlich, L.F. Molecular Determinants of Cancer Therapy Resistance to HDAC Inhibitor-Induced Autophagy. Cancers 2020, 12, 109. https://doi.org/10.3390/cancers12010109
Mrakovcic M, Fröhlich LF. Molecular Determinants of Cancer Therapy Resistance to HDAC Inhibitor-Induced Autophagy. Cancers. 2020; 12(1):109. https://doi.org/10.3390/cancers12010109
Chicago/Turabian StyleMrakovcic, Maria, and Leopold F. Fröhlich. 2020. "Molecular Determinants of Cancer Therapy Resistance to HDAC Inhibitor-Induced Autophagy" Cancers 12, no. 1: 109. https://doi.org/10.3390/cancers12010109
APA StyleMrakovcic, M., & Fröhlich, L. F. (2020). Molecular Determinants of Cancer Therapy Resistance to HDAC Inhibitor-Induced Autophagy. Cancers, 12(1), 109. https://doi.org/10.3390/cancers12010109