Autotaxin Implication in Cancer Metastasis and Autoimunne Disorders: Functional Implication of Binding Autotaxin to the Cell Surface


Abstract
1. Introduction
2. Autotaxin/LysoPLD Activity and Cancer Progression
3. Pharmacological Inhibition of ATX/LysoPLD Activity in Cancer Models
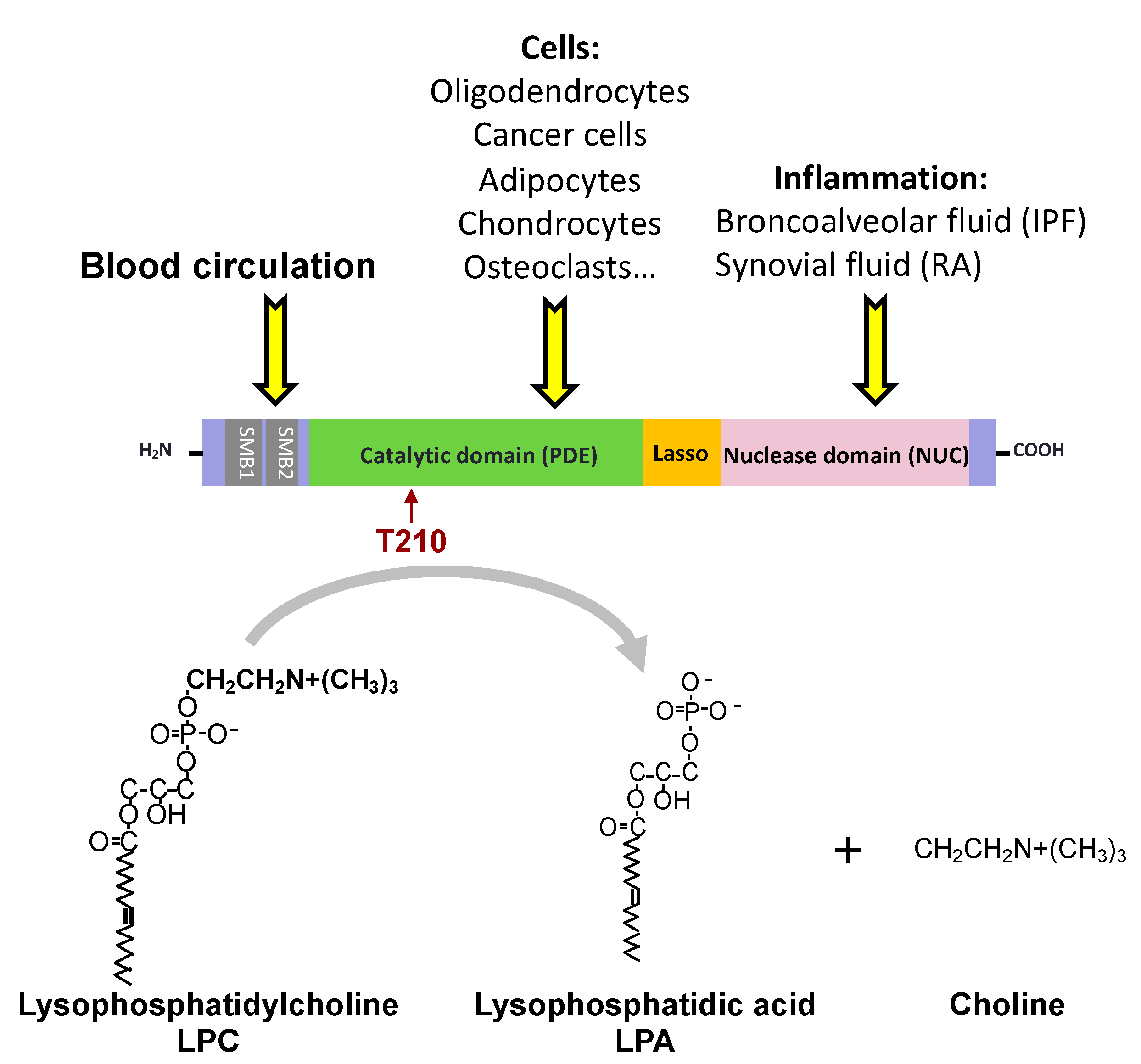
4. ATX Binding to the Cell Surface May Have Functional Impacts on Cell Biology and Pathophysiology
5. Inflammation-Induced Bone Loss is A Pathological Process Controlled by ATX Autocrine Action on Osteoclasts
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar]
- Nam, S.W.; Clair, T.; Campo, C.K.; Lee, H.Y.; Liotta, L.A.; Stracke, M.L. Autotaxin (ATX), a potent tumor motogen, augments invasive and metastatic potential of ras-transformed cells. Oncogene 2000, 19, 241–247. [Google Scholar] [CrossRef]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef]
- Aoki, J.; Taira, A.; Takanezawa, Y.; Kishi, Y.; Hama, K.; Kishimoto, T.; Mizuno, K.; Saku, K.; Taguchi, R.; Arai, H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 2002, 277, 48737–48744. [Google Scholar] [CrossRef]
- Fukushima, N.; Kimura, Y.; Chun, J. A single receptor encoded by vzg-1/lpA1/edg-2 couples to G proteins and mediates multiple cellular responses to lysophosphatidic acid. Proc. Natl. Acad. Sci. USA 1998, 95, 6151–6156. [Google Scholar] [CrossRef]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef]
- Ishii, I.; Contos, J.J.; Fukushima, N.; Chun, J. Functional comparisons of the lysophosphatidic acid receptors, LP(A1)/VZG-1/EDG-2, LP(A2)/EDG-4, and LP(A3)/EDG-7 in neuronal cell lines using a retrovirus expression system. Mol. Pharmacol. 2000, 58, 895–902. [Google Scholar] [CrossRef]
- Bandoh, K.; Aoki, J.; Taira, A.; Tsujimoto, M.; Arai, H.; Inoue, K. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Lett. 2000, 478, 159–165. [Google Scholar] [CrossRef]
- Yanagida, K.; Masago, K.; Nakanishi, H.; Kihara, Y.; Hamano, F.; Tajima, Y.; Taguchi, R.; Shimizu, T.; Ishii, S. Identification and Characterization of a Novel Lysophosphatidic Acid Receptor, p2y5/LPA6. J. Biol. Chem. 2009, 284, 17731–17741. [Google Scholar] [CrossRef]
- Lee, C.W.; Rivera, R.; Dubin, A.E.; Chun, J. LPA(4)/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing G(s)-, G(q)/G(i)-mediated calcium signaling and G(12/13)-mediated Rho activation. J. Biol. Chem. 2007, 282, 4310–4317. [Google Scholar] [CrossRef] [PubMed]
- Kehlen, A.; Englert, N.; Seifert, A.; Klonisch, T.; Dralle, H.; Langner, J.; Hoang-Vu, C. Expression, regulation and function of autotaxin in thyroid carcinomas. Int. J. Cancer. 2004, 109, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Kehlen, A.; Lauterbach, R.; Santos, A.N.; Thiele, K.; Kabisch, U.; Weber, E.; Riemann, D.; Langner, J. IL-1 beta- and IL-4-induced down-regulation of autotaxin mRNA and PC-1 in fibroblast-like synoviocytes of patients with rheumatoid arthritis (RA). Clin. Exp. Immunol. 2001, 123, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kime, C.; Sakaki-Yumoto, M.; Goodrich, L.; Hayashi, Y.; Sami, S.; Derynck, R.; Asahi, M.; Panning, B.; Yamanaka, S.; Tomoda, K. Autotaxin-mediated lipid signaling intersects with LIF and BMP signaling to promote the naive pluripotency transcription factor program. Proc. Natl. Acad. Sci. USA 2016, 113, 12478–12483. [Google Scholar] [CrossRef]
- Flammier, S.; Peyruchaud, O.; Bourguillault, F.; Duboeuf, F.; Davignon, J.L.; Norman, D.D.; Isaac, S.; Marotte, H.; Tigyi, G.; Machuca-Gayet, I.; et al. Osteoclast-Derived Autotaxin, a Distinguishing Factor for Inflammatory Bone Loss. Arthritis Rheumatol. 2019, 71, 1801–1811. [Google Scholar] [CrossRef]
- Wu, J.M.; Xu, Y.; Skill, N.J.; Sheng, H.; Zhao, Z.; Yu, M.; Saxena, R.; Maluccio, M.A. Autotaxin expression and its connection with the TNF-alpha-NF-kappaB axis in human hepatocellular carcinoma. Mol. Cancer 2010, 9, 71. [Google Scholar] [CrossRef]
- Zirn, B.; Samans, B.; Wittmann, S.; Pietsch, T.; Leuschner, I.; Graf, N.; Gessler, M. Target genes of the WNT/beta-catenin pathway in Wilms tumors. Genes Chromosomes Cancer 2006, 45, 565–574. [Google Scholar] [CrossRef]
- Chen, M.; O’Connor, K.L. Integrin alpha6beta4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast carcinoma cells. Oncogene 2005, 24, 5125–5130. [Google Scholar] [CrossRef]
- Leclerc, N.; Luppen, C.A.; Ho, V.V.; Nagpal, S.; Hacia, J.G.; Smith, E.; Frenkel, B. Gene expression profiling of glucocorticoid-inhibited osteoblasts. J. Mol. Endocrinol. 2004, 33, 175–193. [Google Scholar] [CrossRef]
- Dufner-Beattie, J.; Lemons, R.S.; Thorburn, A. Retinoic acid-induced expression of autotaxin in N-myc-amplified neuroblastoma cells. Mol. Carcinog. 2001, 30, 181–189. [Google Scholar] [CrossRef]
- Black, E.J.; Clair, T.; Delrow, J.; Neiman, P.; Gillespie, D.A. Microarray analysis identifies Autotaxin, a tumour cell motility and angiogenic factor with lysophospholipase D activity, as a specific target of cell transformation by v-Jun. Oncogene 2004, 23, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P.; Tarnus, E.; Gres, S.; Valet, P.; Saulnier-Blache, J.S. Secretion and lysophospholipase D activity of autotaxin by adipocytes are controlled by N-glycosylation and signal peptidase. Biochim. Biophys. Acta 2007, 1771, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Andries, M.; Vekemans, K.; Vanbilloen, H.; Verbruggen, A.; Bollen, M. Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells. Cancer Lett. 2009, 284, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.J.; Selim, S.; Salous, A.; Smyth, S.S. Blood Relatives: Dynamic Regulation of Bioactive Lysophosphatidic acid and Sphingosine-1- Phosphate Metabolism in the Circulation. Trends Cardiovasc. Med. 2009, 19, 135–140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tigyi, G.; Miledi, R. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem. 1992, 267, 21360–21367. [Google Scholar] [PubMed]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef]
- Masuda, A.; Nakamura, K.; Izutsu, K.; Igarashi, K.; Ohkawa, R.; Jona, M.; Higashi, K.; Yokota, H.; Okudaira, S.; Kishimoto, T.; et al. Serum autotaxin measurement in haematological malignancies: A promising marker for follicular lymphoma. Br. J. Haematol. 2008, 143, 60–70. [Google Scholar] [CrossRef]
- Nakai, Y.; Ikeda, H.; Nakamura, K.; Kume, Y.; Fujishiro, M.; Sasahira, N.; Hirano, K.; Isayama, H.; Tada, M.; Kawabe, T.; et al. Specific increase in serum autotaxin activity in patients with pancreatic cancer. Clin. Biochem. 2011, 44, 576–581. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Z.; Xu, S.; Ni, L.; Wang, X. Expression of autotaxin mRNA in human hepatocellular carcinoma. Chin. Med. J. 1999, 112, 330–332. [Google Scholar]
- Tigyi, G.J.; Yue, J.; Norman, D.D.; Szabo, E.; Balogh, A.; Balazs, L.; Zhao, G.; Lee, S.C. Regulation of tumor cell—Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv. Biol. Regul. 2019, 71, 183–193. [Google Scholar] [CrossRef]
- Boucharaba, A.; Serre, C.-M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.-C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Investig. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Ko, Y.M.; Tang, X.; Dewald, J.; Lopez-Campistrous, A.; Zhao, Y.Y.; Lai, R.; Curtis, J.M.; Brindley, D.N.; McMullen, T.P.W. Autotaxin is an inflammatory mediator and therapeutic target in thyroid cancer. Endocr.-Relat. Cancer 2015, 22, 593–607. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Dewald, J.; Dong, W.F.; Mackey, J.R.; Hemmings, D.G.; McMullen, T.P.; Brindley, D.N. Tumor-induced inflammation in mammary adipose tissue stimulates a vicious cycle of autotaxin expression and breast cancer progression. FASEB J. 2015, 29, 3990–4000. [Google Scholar] [CrossRef]
- Leblanc, R.; Peyruchaud, O. New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp. Cell Res. 2015, 333, 183–189. [Google Scholar] [CrossRef]
- Benesch, M.G.K.; Yang, Z.; Tang, X.; Meng, G.; Brindley, D.N. Lysophosphatidate Signaling: The Tumor Microenvironment’s New Nemesis. Trends Cancer 2017, 3, 748–752. [Google Scholar] [CrossRef]
- Benesch, M.G.; MacIntyre, I.T.; McMullen, T.P.; Brindley, D.N. Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation. Cancers 2018, 10, 73. [Google Scholar] [CrossRef]
- David, M.; Wannecq, E.; Descotes, F.; Jansen, S.; Deux, B.; Ribeiro, J.; Serre, C.M.; Gres, S.; Bendriss-Vermare, N.; Bollen, M.; et al. Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. PLoS ONE 2010, 5, e9741. [Google Scholar] [CrossRef]
- Vidot, S.; Witham, J.; Agarwal, R.; Greenhough, S.; Bamrah, H.S.; Tigyi, G.J.; Kaye, S.B.; Richardson, A. Autotaxin delays apoptosis induced by carboplatin in ovarian cancer cells. Cell. Signal. 2010, 22, 926–935. [Google Scholar] [CrossRef]
- Mazzocca, A.; Schönauer, L.M.; De Nola, R.; Lippolis, A.; Marrano, T.; Loverro, M.; Sabbà, C.; Di Naro, E. Autotaxin is a novel molecular identifier of type I endometrial cancer. Med. Oncol. 2018, 35, 157. [Google Scholar] [CrossRef]
- Shao, Y.; Yu, Y.; He, Y.; Chen, Q.; Liu, H. Serum ATX as a novel biomarker for breast cancer. Medicine 2019, 98, e14973. [Google Scholar] [CrossRef]
- Magkrioti, C.; Oikonomou, N.; Kaffe, E.; Mouratis, M.A.; Xylourgidis, N.; Barbayianni, I.; Megadoukas, P.; Harokopos, V.; Valavanis, C.; Chun, J.; et al. The autotaxin—Lysophosphatidic acid axis promotes lung carcinogenesis. Cancer Res. 2018, 78, 3634–3644. [Google Scholar] [CrossRef]
- van Meeteren, L.A.; Ruurs, P.; Christodoulou, E.; Goding, J.W.; Takakusa, H.; Kikuchi, K.; Perrakis, A.; Nagano, T.; Moolenaar, W.H. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem. 2005, 280, 21155–21161. [Google Scholar] [CrossRef]
- Ferry, G.; Moulharat, N.; Pradere, J.P.; Desos, P.; Try, A.; Genton, A.; Giganti, A.; Bleucher-Gaudin, M.; Lonchampt, M.; Bertrand, M.; et al. S32826: A nanomolar inhibitor of autotaxin. Discovery, synthesis and applications as a pharmacological tool. J. Pharmacol. Exp. Ther. 2008, 327, 809–819. [Google Scholar] [CrossRef]
- Gupte, R.; Patil, R.; Liu, J.; Wang, Y.; Lee, S.C.; Fujiwara, Y.; Fells, J.; Bolen, A.L.; Emmons-Thompson, K.; Yates, C.R.; et al. Benzyl and naphthalene methylphosphonic acid inhibitors of autotaxin with anti-invasive and anti-metastatic activity. ChemMedChem 2011, 6, 922–935. [Google Scholar] [CrossRef]
- Gotoh, M.; Fujiwara, Y.; Yue, J.; Liu, J.; Lee, S.; Fells, J.; Uchiyama, A.; Murakami-Murofushi, K.; Kennel, S.; Wall, J.; et al. Controlling cancer through the autotaxin-lysophosphatidic acid receptor axis. Biochem. Soc. Trans. 2012, 40, 31–36. [Google Scholar] [CrossRef]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-Pierce, E.; Cortes-Burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S.; et al. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010, 334, 310–317. [Google Scholar] [CrossRef]
- Saga, H.; Ohhata, A.; Hayashi, A.; Katoh, M.; Maeda, T.; Mizuno, H.; Takada, Y.; Komichi, Y.; Ota, H.; Matsumura, N.; et al. A Novel Highly Potent Autotaxin/ENPP2 Inhibitor Produces Prolonged Decreases in Plasma Lysophosphatidic Acid Formation in Vivo and Regulates Urethral Tension. PLoS ONE 2014, 9, e93230. [Google Scholar] [CrossRef]
- Bhave, S.R.; Dadey, D.Y.; Karvas, R.M.; Ferraro, D.J.; Kotipatruni, R.P.; Jaboin, J.J.; Hallahan, A.N.; Dewees, T.A.; Linkous, A.G.; Hallahan, D.E.; et al. Autotaxin Inhibition with PF-8380 Enhances the Radiosensitivity of Human and Murine Glioblastoma Cell Lines. Front. Oncol. 2013, 3, 236. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.; et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef]
- Maher, T.M.; Kreuter, M.; Lederer, D.J.; Brown, K.K.; Wuyts, W.; Verbruggen, N.; Stutvoet, S.; Fieuw, A.; Ford, P.; Abi-Saab, W.; et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir. Res. 2019, 6, e000422. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wuest, M.; Benesch, M.G.; Dufour, J.; Zhao, Y.; Curtis, J.M.; Monjardet, A.; Heckmann, B.; Murray, D.; Wuest, F.; et al. Inhibition of autotaxin with GLPG1690 increases the efficacy of radiotherapy and chemotherapy in a mouse model of breast cancer. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, X.; Gajewiak, J.; Tsukahara, R.; Fujiwara, Y.; Liu, J.; Fells, J.I.; Perygin, D.; Parrill, A.L.; Tigyi, G.; et al. Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor reduces breast cancer cell migration in vitro and causes tumor regression in vivo. Cancer Res. 2009, 69, 5441–5449. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, S.M.; Thotala, D.K.; Linkous, A.G.; Hu, R.; Leahy, K.M.; Yazlovitskaya, E.M.; Hallahan, D.E. Autotaxin and LPA receptors represent potential molecular targets for the radiosensitization of murine glioma through effects on tumor vasculature. PLoS ONE 2011, 6, e22182. [Google Scholar] [CrossRef]
- Iyer, P.; Lalane, R., 3rd; Morris, C.; Challa, P.; Vann, R.; Rao, P.V. Autotaxin-lysophosphatidic acid axis is a novel molecular target for lowering intraocular pressure. PLoS ONE 2012, 7, e42627. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Hwang, S.H.; Lee, B.H.; Kim, H.J.; Cho, H.J.; Shin, H.C.; Im, K.S.; Choi, S.H.; Shin, T.J.; Lee, S.M.; Nam, S.W.; et al. Suppression of metastasis of intravenously-inoculated B16/F10 melanoma cells by the novel ginseng-derived ingredient, gintonin: Involvement of autotaxin inhibition. Int. J. Oncol. 2013, 42, 317–326. [Google Scholar] [CrossRef][Green Version]
- Peyruchaud, O.; David, M.; MacDonald, T.L.; Lynch, K.R. Lysophosphatidic Acid (LPA) Signaling in Bone Cancer. In Lysophospholipid Receptors; Chun, J., Hla, T., Moolenaar, W., Speigel, S., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; pp. 627–640. [Google Scholar]
- Tang, X.; Benesch, M.G.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef]
- Kanda, H.; Newton, R.; Klein, R.; Morita, Y.; Gunn, M.D.; Rosen, S.D. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 2008, 9, 415–423. [Google Scholar] [CrossRef]
- Pamuklar, Z.; Federico, L.; Liu, S.; Umezu-Goto, M.; Dong, A.; Panchatcharam, M.; Fulerson, Z.; Berdyshev, E.; Natarajan, V.; Fang, X.; et al. Autotaxin/lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J. Biol. Chem. 2009, 284, 7385–7394. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Kooi, C.V.; Shah, P.; Charnigo, R.; Huang, C.; Smyth, S.S.; Morris, A.J. Integrin-mediated cell surface recruitment of autotaxin promotes persistent directional cell migration. FASEB J. 2014, 28, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef] [PubMed]
- Houben, A.J.; van Wijk, X.M.; van Meeteren, L.A.; van Zeijl, L.; van de Westerlo, E.M.; Hausmann, J.; Fish, A.; Perrakis, A.; van Kuppevelt, T.H.; Moolenaar, W.H. The polybasic insertion in autotaxin alpha confers specific binding to heparin and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2013, 288, 510–519. [Google Scholar] [CrossRef]
- Leblanc, R.; Sahay, D.; Houssin, A.; Machuca-Gayet, I.; Peyruchaud, O. Autotaxin-beta interaction with the cell surface via syndecan-4 impacts on cancer cell proliferation and metastasis. Oncotarget 2018, 9, 33170–33185. [Google Scholar] [CrossRef]
- Jethwa, S.A.; Leah, E.J.; Zhang, Q.; Bright, N.A.; Oxley, D.; Bootman, M.D.; Rudge, S.A.; Wakelam, M.J. Exosomes bind to autotaxin and act as a physiological delivery mechanism to stimulate LPA receptor signalling in cells. J. Cell Sci. 2016, 129, 3948–3957. [Google Scholar] [CrossRef]
- Cohen, G.; Gossec, L.; Dougados, M.; Cantagrel, A.; Goupille, P.; Daures, J.P.; Rincheval, N.; Combe, B. Radiological damage in patients with rheumatoid arthritis on sustained remission. Ann. Rheum. Dis. 2007, 66, 358–363. [Google Scholar] [CrossRef]
- Redlich, K.; Hayer, S.; Ricci, R.; David, J.-P.; Tohidast-Akrad, M.; Kollias, G.; Steiner, G.; Smolen, J.S.; Wagner, E.F.; Schett, G. Osteoclasts are essential for TNF-{alpha}-mediated joint destruction. J. Clin. Investig. 2002, 110, 1419–1427. [Google Scholar] [CrossRef]
- Molenaar, E.T.; Voskuyl, A.E.; Dinant, H.J.; Bezemer, P.D.; Boers, M.; Dijkmans, B.A. Progression of radiologic damage in patients with rheumatoid arthritis in clinical remission. Arthritis Rheum. 2004, 50, 36–42. [Google Scholar] [CrossRef]
- Brown, A.K.; Conaghan, P.G.; Karim, Z.; Quinn, M.A.; Ikeda, K.; Peterfy, C.G.; Hensor, E.; Wakefield, R.J.; O’Connor, P.J.; Emery, P. An explanation for the apparent dissociation between clinical remission and continued structural deterioration in rheumatoid arthritis. Arthritis Rheum. 2008, 58, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, Y.; Miyabe, C.; Iwai, Y.; Takayasu, A.; Fukuda, S.; Yokoyama, W.; Nagai, J.; Jona, M.; Tokuhara, Y.; Ohkawa, R.; et al. Necessity of lysophosphatidic acid receptor 1 for development of arthritis. Arthritis Rheum. 2013, 65, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Fernandes, M.J.; Prestwich, G.D.; Turgeon, M.; Di Battista, J.; Clair, T.; Poubelle, P.E.; Bourgoin, S.G. Regulation of lysophosphatidic acid receptor expression and function in human synoviocytes: Implications for rheumatoid arthritis? Mol. Pharmacol. 2008, 73, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef]
- David, M.; Machuca-Gayet, I.; Kikuta, J.; Ottewell, P.; Mima, F.; Leblanc, R.; Bonnelye, E.; Ribeiro, J.; Holen, I.; Vales, R.L.; et al. Lysophosphatidic Acid Receptor Type 1 (LPA1) Plays a Functional Role in Osteoclast Differentiation and Bone Resorption Activity. J. Biol. Chem. 2014, 289, 6551–6564. [Google Scholar] [CrossRef]
- Dusaulcy, R.; Daviaud, D.; Pradere, J.P.; Gres, S.; Valet, P.; Saulnier-Blache, J.S. Altered food consumption in mice lacking lysophosphatidic acid receptor-1. J. Physiol. Biochem. 2009, 65, 345–350. [Google Scholar] [CrossRef]
- Contos, J.J.A.; Fukushima, N.; Weiner, J.A.; Kaushal, D.; Chun, J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. USA 2000, 97, 13384–13389. [Google Scholar] [CrossRef]
- Ye, X.; Hama, K.; Contos, J.J.; Anliker, B.; Inoue, A.; Skinner, M.K.; Suzuki, H.; Amano, T.; Kennedy, G.; Arai, H.; et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 2005, 435, 104–108. [Google Scholar] [CrossRef]
- Nishioka, T.; Arima, N.; Kano, K.; Hama, K.; Itai, E.; Yukiura, H.; Kise, R.; Inoue, A.; Kim, S.H.; Solnica-Krezel, L.; et al. ATX-LPA1 axis contributes to proliferation of chondrocytes by regulating fibronectin assembly leading to proper cartilage formation. Sci. Rep. 2016, 6, 23433. [Google Scholar] [CrossRef]
- Panupinthu, N.; Rogers, J.T.; Zhao, L.; Solano-Flores, L.P.; Possmayer, F.; Sims, S.M.; Dixon, S.J. P2 × 7 receptors on osteoblasts couple to production of lysophosphatidic acid: A signaling axis promoting osteogenesis. J. Cell Biol. 2008, 181, 859–871. [Google Scholar] [CrossRef]
- Ferry, G.; Tellier, E.; Try, A.; Gres, S.; Naime, I.; Simon, M.F.; Rodriguez, M.; Boucher, J.; Tack, I.; Gesta, S.; et al. Autotaxin Is Released from Adipocytes, Catalyzes Lysophosphatidic Acid Synthesis, and Activates Preadipocyte Proliferation. Up-Regulated Expression with Adipocyte Differentiation and Obesity. J. Biol. Chem. 2003, 278, 18162–18169. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Heath, D.J.; Vanderkerken, K.; Cheng, X.; Gallagher, O.; Prideaux, M.; Murali, R.; Croucher, P.I. An osteoprotegerin-like peptidomimetic inhibits osteoclastic bone resorption and osteolytic bone disease in myeloma. Cancer Res. 2007, 67, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- McHugh, K.P.; Hodivala-Dilke, K.; Zheng, M.H.; Namba, N.; Lam, J.; Novack, D.; Feng, X.; Ross, F.P.; Hynes, R.O.; Teitelbaum, S.L. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Investig. 2000, 105, 433–440. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Receptors | G Proteins | Cellular Responses |
|---|---|---|
| LPA1/Edg2 | Gi/o, Gq/11, G12,13 | Neurite retraction [6,7], AC inhibition [8], SRE activation [6], increased [Ca2+]i, IP production, MAPK activation [8], stress fiber formation, BrdU incorporation [6], inhibition of apoptosis, arachidonic acid release [8]. |
| LPA2/Edg4 | Gi/o, Gq/11, G12,13 | Neurite retraction [8], AC inhibition [9], SRE activation, increased [Ca2+]i [9], IP production [8], MAPK activation [8], stress fiber formation, BrdU incorporation [6], inhibition of apoptosis, arachidonic acid release [8]. |
| LPA3/Edg7 | Gi/o, Gq/11 | AC inhibition [8], increased [Ca2+]i, IP production, MAPK activation, arachidonic acid release [8]. |
| LPA4/p2y9/GPR23 | Gq/11, G12/13, Gs, (Gi) | AC stimulation, increased [Ca2+]i [10], zif268 activation, neurite retraction, cell aggregation [10], stress fiber formation [11]. |
| LPA5/GPR92/GPR93 | Gq/11, G12/13 | AC stimulation, increased [Ca2+]i, IP production, neurite retraction [7]. |
| LPA6/p2y5 | G12/13, (Gs), (Gi) | CRE activation, neurite retraction, membrane shedding [10]. |
| External Signals | Transcription Factors | Effects | Cell Types | References |
|---|---|---|---|---|
| EGF | nd | Upregulation | Thyroid cancer cells | [12] |
| β-FGF | nd | Upregulation | Thyroid cancer cells | [12] |
| IL-4 | nd | Downregulation | Fibroblast-like synoviocytes | [13] |
| IL-1β | nd | Downregulation | Fibroblast-like synoviocytes | [13] |
| BMP4 | nd | Upregulation | Primed pluripotent stem cells | [14] |
| TNFα | NF-κB | Upregulation | Osteoclasts Transformed fibroblasts | [15] [16] |
| LPS | NF-κB | Upregulation | Osteoclasts | [15] |
| WNT | β-Catenin | Upregulation | Wilms tumors | [17] |
| α6β4 | NFAT1 | Upregulation | MDA-MB-435 cells | [18] |
| Glucocorticoid | nd | Upregulation | MC3T3-E1 cells | [19] |
| Retinoic acid | nd | Upregulation | N-myc-amplified neuroblastoma cells | [20] |
| ------ | v-jun | Upregulation | Chick embryo fibroblasts | [21] |
| Drug Names | Cancer Types | Effects | Ref. |
|---|---|---|---|
| S32826 | Ovarian cancer | Primary tumor growth retardation of OCAR-3 cells at high doses | [44] |
| BrP-LPA | Breast cancer | Primary tumor growth inhibition of MDA-MB-231 cells | [53] |
| BrP-LPA | Glioma | Primary tumor growth delay of GL-261 cells in combination with radiotherapy | [54] |
| BMP-22 | Melanoma | Inhibition of lung metastasis of B16-F10 cells | [45] |
| BMP-30a | Melanoma | Inhibition of lung metastasis of B16-F10 cells | [45] |
| S32826 | nd | Decreased intraocular pressure in Dutch-Belted rabbits (glaucoma) | [55] |
| GWJ-A-23 | nd | Attenuation of idiopathic pulmonary fibrosis induced by bleaomycine treatment | [56] |
| PF-8380 | Glioblastoma | Ameliorates the glioblastoma GL261 cell response to radiotherapy | [49] |
| Gintonin | Melanoma | Inhibition of lung metastasis of B16-F10 cells | [57] |
| VPC8a202 | Breast cancer | Inhibition of lung metastasis of 4T1 cells | [58] |
| ONO-8430506 | Breast cancer | Inhibition of tumor growth and lung metastasis of 4T1 cells | [50] |
| BMP-22 | Breast cancer | Inhibition of bone metastasis of MDA-BO2 cells | [32] |
| GLPG1609 | Current Phase 3 clinical trials for idiopathic pulmonary fibrosis | [51] | |
| GLPG1609 | Breast cancer | Sensitizes primary tumors of 4T1 cells to radiotherapy and chemotherapy | [52] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyruchaud, O.; Saier, L.; Leblanc, R. Autotaxin Implication in Cancer Metastasis and Autoimunne Disorders: Functional Implication of Binding Autotaxin to the Cell Surface. Cancers 2020, 12, 105. https://doi.org/10.3390/cancers12010105
Peyruchaud O, Saier L, Leblanc R. Autotaxin Implication in Cancer Metastasis and Autoimunne Disorders: Functional Implication of Binding Autotaxin to the Cell Surface. Cancers. 2020; 12(1):105. https://doi.org/10.3390/cancers12010105
Chicago/Turabian StylePeyruchaud, Olivier, Lou Saier, and Raphaël Leblanc. 2020. "Autotaxin Implication in Cancer Metastasis and Autoimunne Disorders: Functional Implication of Binding Autotaxin to the Cell Surface" Cancers 12, no. 1: 105. https://doi.org/10.3390/cancers12010105
APA StylePeyruchaud, O., Saier, L., & Leblanc, R. (2020). Autotaxin Implication in Cancer Metastasis and Autoimunne Disorders: Functional Implication of Binding Autotaxin to the Cell Surface. Cancers, 12(1), 105. https://doi.org/10.3390/cancers12010105