Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (ctDNA) Analysis: Complementary Modalities in Clinical Practice

 ,
,
Abstract
1. Introduction
2. Materials and Methods
Methods
3. Results
3.1. Patient Characteristics
3.2. Patient-Level Analysis
3.3. Gene-Level, Driver Mutation, and Actionable Alteration Analysis
3.4. Hotspot Mutational Analysis Concordance
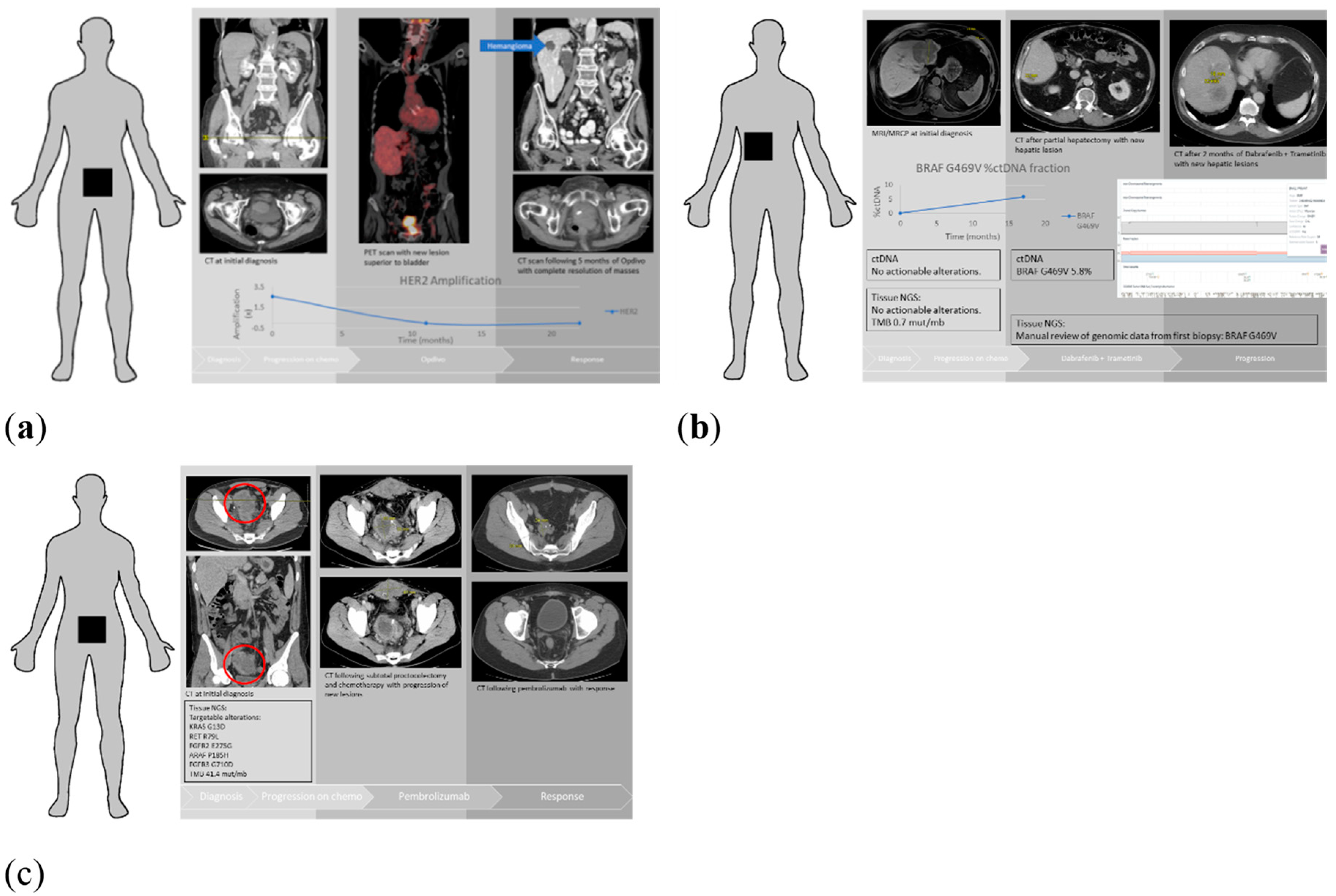
3.5. Patient Examples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Brana, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; O’Day, S.J.; Kitago, M.; Amersi, F.; Kuo, C.; Kim, J.; Wang, H.; Hoon, D.S.B. Utility of Circulating B-RAF DNA Mutation in Serum for Monitoring Melanoma Patients Receiving Biochemotherapy. Clin. Cancer Res. 2007, 13, 2068–2074. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef]
- Kim, S.T.; Banks, K.C.; Lee, S.-H.; Kim, K.; Park, J.O.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Lanman, R.B.; et al. Prospective Feasibility Study for Using Cell-Free Circulating Tumor DNA–Guided Therapy in Refractory Metastatic Solid Cancers: An Interim Analysis. JCO Precis. Oncol. 2017, 1, 1–15. [Google Scholar] [CrossRef]
- Toor, O.M.; Ahmed, Z.; Bahaj, W.; Boda, U.; Cummings, L.S.; McNally, M.E.; Kennedy, K.F.; Pluard, T.J.; Hussain, A.; Subramanian, J.; et al. Correlation of Somatic Genomic Alterations Between Tissue Genomics and ctDNA Employing Next-Generation Sequencing: Analysis of Lung and Gastrointestinal Cancers. Mol. Cancer Ther. 2018, 17, 1123–1132. [Google Scholar] [CrossRef]
- Barata, P.C.; Koshkin, V.S.; Funchain, P.; Sohal, D.; Pritchard, A.; Klek, S.; Adamowicz, T.; Gopalakrishnan, D.; Garcia, J.; Rini, B.; et al. Next-generation sequencing (NGS) of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: A pilot assessment of concordance. Ann. Oncol. 2017, 28, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Patel, S.P.; Harismendy, O.; Ikeda, M.; Parker, B.A.; et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clin. Cancer Res. 2016, 22, 5497–5505. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristonfailli, M. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.W.; Gill, D.M.; Maughan, B.; Agarwal, A.; Arjyal, L.; Gupta, S.; Streeter, J.; Bailey, E.; Pal, S.K.; Agarwal, N. Correlation of genomic alterations assessed by next-generation sequencing (NGS) of tumor tissue DNA and circulating tumor DNA (ctDNA) in metastatic renal cell carcinoma (mRCC): Potential clinical implications. Oncotarget 2017, 8, 33614. [Google Scholar] [CrossRef] [PubMed]
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor–Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, W.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djx118. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.-C.; Covington, K.R.; Chang, D.K.; Donehower, L.A.; Gill, A.J.; Ittmann, M.M.; Creighton, C.J.; Johns, A.L.; Shinbrot, E.; Dewal, N.; et al. Ampullary Cancers Harbor ELF3 Tumor Suppressor Gene Mutations and Exhibit Frequent WNT Dysregulation. Cell Rep. 2016, 14, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Wilson, J.K.; Sedwick, W.D.; Wang, Z.J.; et al. Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Narayan, R.R.; Creasy, J.M.; Goldman, D.A.; Gönen, M.; Kandoth, C.; Kundra, R.; Solid, D.B.; Askan, G.; Klimstra, D.S.; Basturk, O.; et al. Regional differences in gallbladder cancer pathogenesis: Insights from a multi-institutional comparison of tumor mutations. Cancer 2019, 125, 575–585. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Kuderer, N.M.; Burton, K.A.; Blau, S.; Rose, A.L.; Parker, S.; Lyman, G.H.; Blau, C.A. Comparison of 2 Commercially Available Next-Generation Sequencing Platforms in Oncology. JAMA Oncol. 2017, 3, 996. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.D.; Kalyan, A.; Taxter, T.J.; Wehbe, F.; Kircher, S.M.; Nimeiri, H.S.; Mulcahy, M.F.; Benson, A.B.; Cristofanilli, M.; Giles, F.J. Concordance of mutations identified using circulating tumor DNA (ctDNA) compared to tissue based next generation sequencing (NGS) in gastrointestinal malignancies: A single institution experience. J. Clin. Oncol. 2017, 35, e23023. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Pedrero, M.; Lefebvre, C.; Marabelle, A.; Soria, J.C.; Postel-Vinay, S. Mutational Landscape and Sensitivity to Immune Checkpoint Blockers. Clin. Cancer Res. 2016, 22, 4309–4321. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Danilova, L.; Wang, H.; Sunshine, J.; Kaunitz, G.J.; Cottrell, T.R.; Xu, H.; Esandrio, J.; Anders, R.A.; Cope, L.; Pardoll, D.M.; et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc. Natl. Acad. Sci. USA 2016, 113, E7769–E7777. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Shatsky, R.; Parker, B.A.; Bui, N.Q.; Helsten, T.; Schwab, R.B.; Boles, S.G.; Kurzrock, R. Next-Generation Sequencing of Tissue and Circulating Tumor DNA: The UC San Diego Moores Center for Personalized Cancer Therapy Experience with Breast Malignancies. Mol. Cancer Ther. 2019, 18, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Schwaederlé, M.C.; Fanta, P.T.; Okamura, R.; Leichman, L.; Lippman, S.M.; Lanman, R.B.; Raymond, V.M.; Talasaz, A.; Kurzrock, R. Genomic Assessment of Blood-Derived Circulating Tumor DNA in Patients with Colorectal Cancers: Correlation With Tissue Sequencing, Therapeutic Response, and Survival. JCO Precis. Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef]
- Yang, N.; Li, Y.; Liu, Z.; Qin, H.; Du, D.; Cao, X.; Cao, X.; Li, J.; Li, D.; Jiang, B.; et al. The characteristics of ctDNA reveal the high complexity in matching the corresponding tumor tissues. BMC Cancer 2018, 18, 319. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef]
- Stockley, T.L.; Oza, A.M.; Berman, H.K.; Leighl, N.B.; Knox, J.J.; Shepherd, F.A.; Chen, E.X.; Krzyzanowska, M.K.; Dhani, N.; Joshua, A.M.; et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: The Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016, 8, 109. [Google Scholar] [CrossRef]
- Shahbandi, A.; Jackson, J.G. Analysis across multiple tumor types provides no evidence that mutant p53 exerts dominant negative activity. NPJ Precis. Oncol. 2019, 3, 1. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Patients Characteristics | Total Number of Patients (n = 64) |
|---|---|
| Median age at diagnosis (years) | 66 |
| Sex: | |
| Male | 22 (34.4%) |
| Female | 42 (65.6%) |
| Type of cancer: | |
| GI cancer | 20 (31.2%) |
| Lung cancer | 19 (29.7%) |
| Breast cancer | 12 (18.7%) |
| Other malignancies | 13 (20.3%) |
| Median time between tissue biopsy and blood specimen collection | 20.5 months |
| Biopsy site | |
| Primary tumor | 38 (59%) |
| Metastatic site | 26 (41%) |
| Tumor stage | |
| Stage III | 8 (12.5%) |
| Stage IV | 56 (87.5%) |
| Variable | Patient Level (%) | Gene Level | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| p-value | All Alterations * (%) | p-value | Driver Alterations (%) | p-value | Targetable Alterations (%) | p-value | Hotspot Alterations (%) | p-value | ||
| Tumor type | ||||||||||
| All tumor types | 58 | 16 | 9 | 45 | 34 | |||||
| Breast carcinoma | 83 | 20 | 10 | 37 | 36 | - | ||||
| Lung carcinoma | 68 | 22 | 12 | 44 | 43 | |||||
| GI malignancies | 45 | - | 15 | - | 12 | - | 26 | - | 35 | |
| Tumor mutational burden (mutations/megabase) | ||||||||||
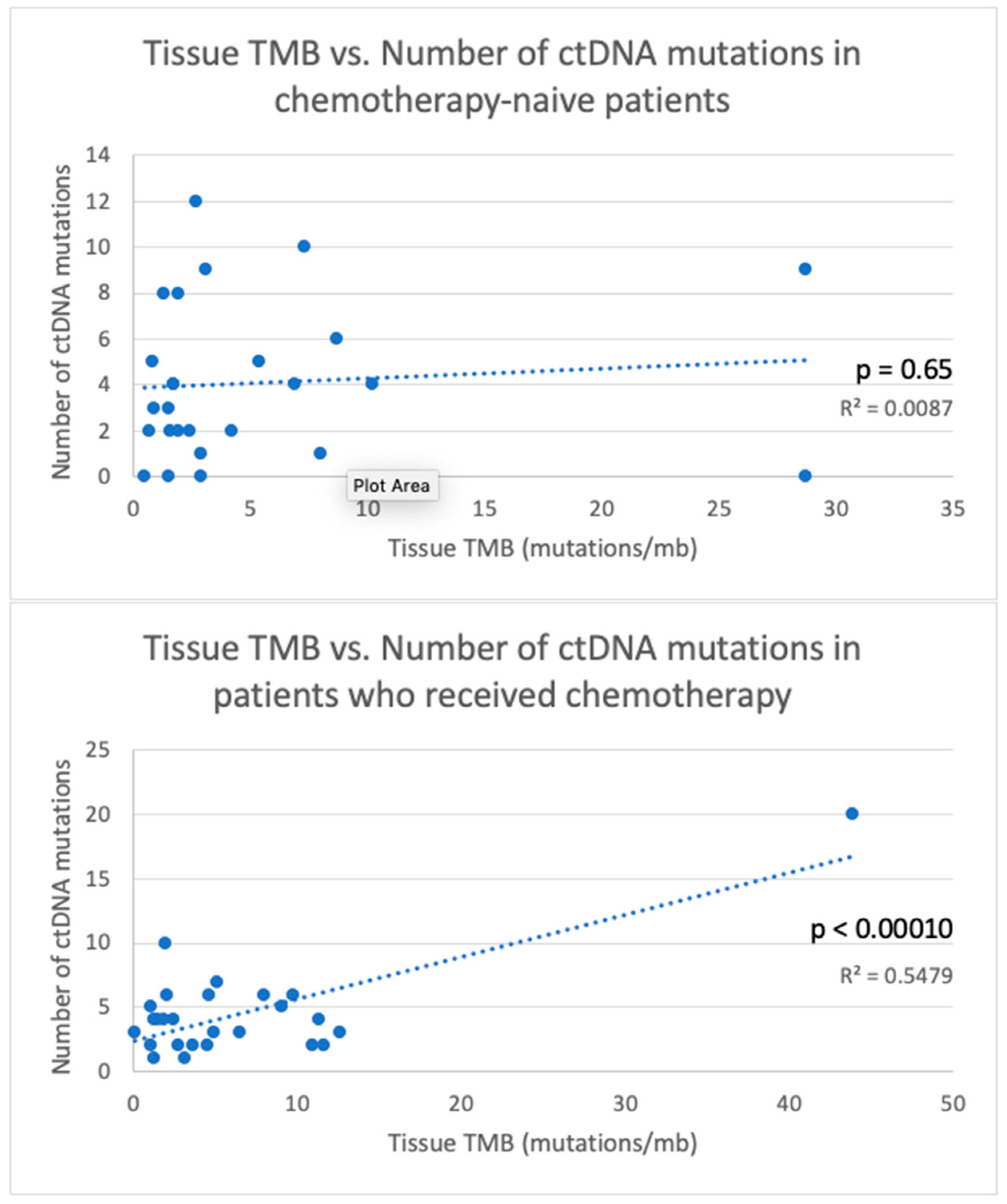
| TMB < 2 | 36 | 0.017 | 13 | 0.40 | 10 | 0.72 | 38 | 0.27 | 21 | 0.013 |
| TMB ≥ 2 | 69 | 17 | 9 | 20 | 39 | |||||
| Chemotherapy status | ||||||||||
| Received chemotherapy before testing | 78 | 0.013 | 21 | 9 | 29 | 43 | 0.15 | |||
| Chemotherapy-naïve | 43 | 11 | 0.014 | 7 | 0.31 | 9.5 | 0.10 | 26 | ||
| Interval between tissue NGS and ctDNA (days) | ||||||||||
| <90 | 55 | 0.43 | 16 | 0.71 | 9 | 0.93 | 37 | 0.79 | 32 | 0.63 |
| ≥90 | 65 | 17 | 9 | 32 | 38 | |||||
| Biopsy site | ||||||||||
| Primary site | 48 | 14 | 10 | 27 | 33 | 1.0 | ||||
| Metastatic site | 64 | 0.20 | 17 | 0.50 | 9 | 0.71 | 23 | 0.74 | 35 | |
| Number of metastatic lesions | ||||||||||
| 1 metastatic lesion present | 53 | 15 | 10 | 13 | 39 | |||||
| >1 metastatic lesion present | 55 | 0.94 | 17 | 0.52 | 9 | 0.67 | 27 | 0.48 | 32 | 0.63 |
| Variable | Total TP53 HS Mutations | Concordant TP53 HS Mutations | Percent Concordance of TP53 HS Mutations | Tissue NGS | Tissue NGS (%) | ctDNA | ctDNA (%) |
|---|---|---|---|---|---|---|---|
| Tumor types | |||||||
| All tumor types | 48 | 14 | 29.17% | 15 | 31.25% | 19 | 39.58% |
| Breast carcinoma | 8 | 2 | 25.00% | 3 | 37.50% | 3 | 37.50% |
| Lung carcinoma | 16 | 6 | 37.50% | 3 | 18.75% | 7 | 43.75% |
| GI malignancies | 13 | 4 | 30.77% | 4 | 30.77% | 5 | 38.46% |
| Other malignancies | 11 | 2 | 18.18% | 5 | 45.45% | 4 | 36.36% |
| Chemotherapy status | |||||||
| Received chemotherapy before testing | 22 | 7 | 31.82% | 6 | 27.27% | 9 | 40.91% |
| Chemotherapy-naïve | 20 | 5 | 25.00% | 7 | 35.00% | 8 | 40.00% |
| Biopsy site | |||||||
| Primary site | 22 | 7 | 31.82% | 7 | 31.82% | 8 | 36.36% |
| Metastatic site | 26 | 7 | 26.92% | 8 | 30.77% | 11 | 42.31% |
| Interval between tissue NGS and ctDNA (days) | |||||||
| <90 | 34 | 11 | 32.35% | 10 | 29.41% | 13 | 38.24% |
| ≥90 | 14 | 3 | 21.43% | 5 | 35.71% | 6 | 42.86% |
| Variables | Total HS Mutations | Concordant HS Mutations | Percent Concordance for HS Mutations | Most Frequent HS Mutation | Most Frequent Concordant HS Mutation | Most Frequent Discordant HS Mutation |
|---|---|---|---|---|---|---|
| Tumor type | ||||||
| All tumor types | 91 | 31 | 34.07% | KRAS G12/G13 (15) TP53 R175 (4) KRAS G12/G13 (6) KRAS G12 (7) KRAS G12 (2) TP53 R273 (2) | KRAS G12/G13 (9) TP53 R175 (2) KRAS G12/G13 (5) KRAS G12 (4) BRAF V600 TP53 E285 TP53 S241 | KRAS G12/G13 (6) |
| Breast carcinoma | 14 | 5 | 35.71% | |||
| Lung carcinoma | 28 | 12 | 42.86% | |||
| GI malignancies | 31 | 11 | 35.48% | |||
| Other malignancies | 18 | 3 | 16.67% | |||
| Chemotherapy status | ||||||
| Received chemotherapy before testing | 35 | 15 | 42.86% | KRAS G12 (5) | KRAS G12 (5) | TP53 R248 (2) TP53 Y220 (2) |
| Chemotherapy-naïve | 46 | 12 | 26.09% | KRAS G12/G13 (9) | KRAS G12 (3) | KRAS G12/G13 (6) |
| Biopsy site | ||||||
| Primary site | 36 | 12 | 33.33% | KRAS G12 (6) | KRAS G12 (4) | TP53 R213 (3) TP53 Y220 (3) |
| Metastatic site | 55 | 19 | 34.55% | KRAS G12/G13 (10) | KRAS G12/G13 (5) | KRAS G12/G13 (5) |
| Interval between tissue NGS and ctDNA (days) | ||||||
| <90 | 65 | 21 | 32.31% | KRAS G12/G13 (11) | KRAS G12 (5) | KRAS G12/G13 (6) |
| ≥90 | 26 | 10 | 38.46% | KRAS G12/G13 (4) | KRAS G12/G13 (4) | TP53 R213 (3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imperial, R.; Nazer, M.; Ahmed, Z.; Kam, A.E.; Pluard, T.J.; Bahaj, W.; Levy, M.; Kuzel, T.M.; Hayden, D.M.; Pappas, S.G.; et al. Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (ctDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers 2019, 11, 1399. https://doi.org/10.3390/cancers11091399
Imperial R, Nazer M, Ahmed Z, Kam AE, Pluard TJ, Bahaj W, Levy M, Kuzel TM, Hayden DM, Pappas SG, et al. Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (ctDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers. 2019; 11(9):1399. https://doi.org/10.3390/cancers11091399
Chicago/Turabian StyleImperial, Robin, Marjan Nazer, Zaheer Ahmed, Audrey E. Kam, Timothy J. Pluard, Waled Bahaj, Mia Levy, Timothy M. Kuzel, Dana M. Hayden, Sam G. Pappas, and et al. 2019. "Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (ctDNA) Analysis: Complementary Modalities in Clinical Practice" Cancers 11, no. 9: 1399. https://doi.org/10.3390/cancers11091399
APA StyleImperial, R., Nazer, M., Ahmed, Z., Kam, A. E., Pluard, T. J., Bahaj, W., Levy, M., Kuzel, T. M., Hayden, D. M., Pappas, S. G., Subramanian, J., & Masood, A. (2019). Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (ctDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers, 11(9), 1399. https://doi.org/10.3390/cancers11091399