Significance of BRAF Kinase Inhibitors for Melanoma Treatment: From Bench to Bedside


Abstract
1. Introduction
2. Methods
Search Strategy for the Literature Search
3. Pre-Clinical Investigations of BRAF Kinase Inhibitors
4. Clinical Trials of BRAF Kinase Inhibitors in the Treatment of Melanoma
4.1. Phase I Studies
4.2. Phase II Studies
4.3. Phase III Studies
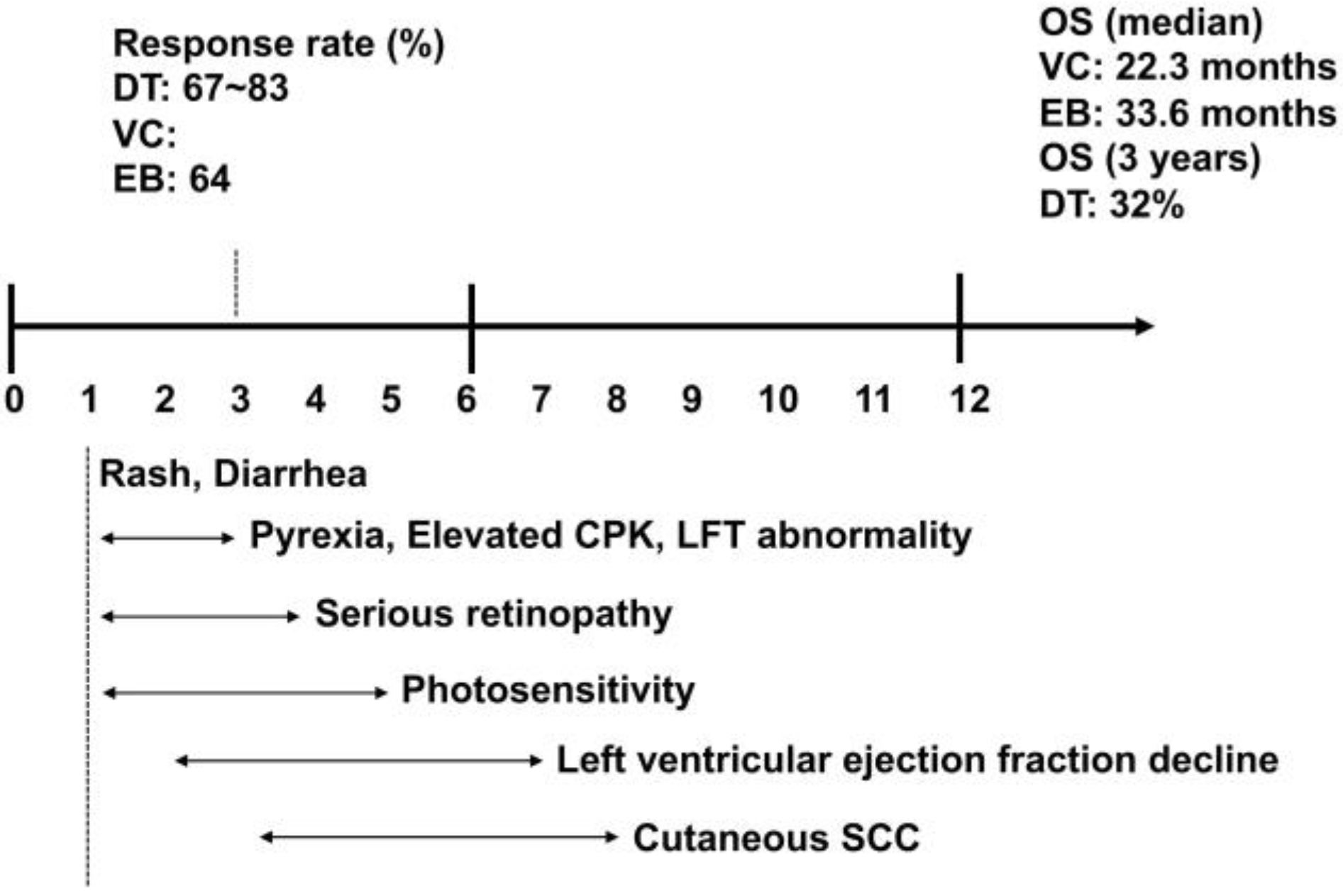
4.4. Adverse Events
5. Future Perspective
Funding
Conflicts of Interest
References
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Grob, J.J.; Amonkar, M.M.; Karaszewska, B.; Schachter, J.; Dummer, R.; Mackiewicz, A.; Stroyakovskiy, D.; Drucis, K.; Grange, F.; Chiarion-Sileni, V.; et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): Results of a phase 3, open-label, randomised trial. Lancet Oncol. 2015, 16, 1389–1398. [Google Scholar] [PubMed]
- Grob, J.J.; Amonkar, M.M.; Martin-Algarra, S.; Demidov, L.V.; Goodman, V.; Grotzinger, K.; Haney, P.; Kämpgen, E.; Karaszewska, B.; Mauch, C.; et al. Patient perception of the benefit of a BRAF inhibitor in metastatic melanoma: Quality-of-life analyses of the BREAK-3 study comparing dabrafenib with dacarbazine. Ann. Oncol. 2014, 25, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Hidaka, T.; Kambayashi, Y.; Aiba, S. BRAF kinase inhibitors for treatment of melanoma: Developments from early-stage animal studies to Phase II clinical trials. Expert Opin. Investig. Drugs 2019, 28, 143–148. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Sakaizawa, K.; Ashida, A.; Uchiyama, A.; Ito, T.; Fujisawa, Y.; Ogata, D.; Matsushita, S.; Fujii, K.; Fukushima, S.; Shibayama, Y.; et al. Clinical characteristics associated with BRAF, NRAS and KIT mutations in Japanese melanoma patients. J. Dermatol. Sci. 2015, 80, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Saida, T.; Yamamoto, A. Japanese Skin Cancer Society Prognosis and Statistical Investigation Committee. Updated statistical data for malignant melanoma in Japan. Int. J. Clin. Oncol. 2001, 6, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Madore, J.; Strbenac, D.; Vilain, R.; Menzies, A.M.; Yang, J.Y.; Thompson, J.F.; Long, G.V.; Mann, G.J.; Scolyer, R.A.; Wilmott, J.S. PD-L1 Negative Status is Associated with Lower Mutation Burden, Differential Expression of Immune-Related Genes, and Worse Survival in Stage III Melanoma. Clin. Cancer Res. 2016, 22, 3915–3923. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Aspromonte, S.; Zloza, A.; Rabkin, S.D.; Kaufman, H.L. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci. Transl. Med. 2018, 10, eaau0417. [Google Scholar] [CrossRef]
- Atay, C.; Kwak, T.; Lavilla-Alonso, S.; Donthireddy, L.; Richards, A.; Moberg, V.; Pilon-Thomas, S.; Schell, M.; Messina, J.L.; Rebecca, V.W.; et al. BRAF Targeting Sensitizes Resistant Melanoma to Cytotoxic T Cells. Clin. Cancer Res. 2019, 25, 2783–2794. [Google Scholar] [CrossRef]
- Mok, S.; Tsoi, J.; Koya, R.C.; Hu-Lieskovan, S.; West, B.L.; Bollag, G.; Graeber, T.G.; Ribas, A. Inhibition of colony stimulating factor-1 receptor improves antitumor efficacy of BRAF inhibition. BMC Cancer 2015, 15, 356. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.M.; Shabaneh, T.B.; Zhang, P.; Martyanov, V.; Li, Z.; Malik, B.T.; Wood, T.A.; Boni, A.; Molodtsov, A.; Angeles, C.V.; et al. Myeloid Cells That Impair Immunotherapy Are Restored in Melanomas with Acquired Resistance to BRAF Inhibitors. Cancer Res. 2017, 77, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Fujisawa, Y.; Hidaka, T.; Aiba, S. Tumor-associated macrophages: Therapeutic targets for skin cancer. Front. Oncol. 2018, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination with Dacarbazine in Patients with Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Furudate, S.; Fujimura, T.; Kambayashi, Y.; Hidaka, T.; Sato, Y.; Tanita, K.; Hashimoto, A.; Aiba, S. Keratoacanthoma, palmoplantar keratoderma developing from an advanced melanoma patient treated with vemurafenib regressed by the blockade of MEK signaling. J. Dermatol. 2017, 44, e226–e227. [Google Scholar] [CrossRef]
- Corre, S.; Tardif, N.; Mouchet, N.; Leclair, H.M.; Boussemart, L.; Gautron, A.; Bachelot, L.; Perrot, A.; Soshilov, A.; Rogiers, A.; et al. Sustained activation of the Aryl hydrocarbon Receptor transcription factor promotes resistance to BRAF-inhibitors in melanoma. Nat. Commun. 2018, 9, 4775. [Google Scholar] [CrossRef]
- Gupta, R.; Bugide, S.; Wang, B.; Green, M.R.; Johnson, D.B.; Wajapeyee, N. Loss of BOP1 confers resistance to BRAF kinase inhibitors in melanoma by activating MAP kinase pathway. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]
- Thakur, M.D.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Heijkants, R.; Willekens, K.; Schoonderwoerd, M.; Teunisse, A.; Nieveen, M.; Radaelli, E.; Hawinkels, L.; Marine, J.C.; Jochemsen, A. Combined inhibition of CDK and HDAC as a promising therapeutic strategy for both cutaneous and uveal metastatic melanoma. Oncotarget 2017, 9, 6174–6187. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Gunatilake, D.; Beaumont, K.A.; Sharp, D.M.; Tiffen, J.C.; Heinemann, A.; Weninger, W.; Haass, N.K.; Wilmott, J.S.; Madore, J.; et al. HDAC inhibitors restore BRAF-inhibitor sensitivity by altering PI3K and survival signalling in a subset of melanoma. Int. J. Cancer 2018, 142, 1926–1937. [Google Scholar] [CrossRef] [PubMed]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Homet Moreno, B.; Mok, S.; Comin-Anduix, B.; Hu-Lieskovan, S.; Ribas, A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology 2015, 5, e1052212. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Fujimura, T.; Kakizaki, A.; Furudate, S.; Kambayashi, Y.; Aiba, S. Tumor-associated macrophages in skin: How to treat their heterogeneity and plasticity. J. Dermatol. Sci. 2016, 83, 167–173. [Google Scholar] [CrossRef]
- Hajek, E.; Krebs, F.; Bent, R.; Haas, K.; Bast, A.; Steinmetz, I.; Tuettenberg, A.; Grabbe, S.; Bros, M. BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget 2018, 9, 28294–28308. [Google Scholar] [CrossRef]
- Gowrishankar, K.; Snoyman, S.; Pupo, G.M.; Becker, T.M.; Kefford, R.F.; Rizos, H. Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition. J. Investig. Dermatol. 2012, 132, 1850–1859. [Google Scholar] [CrossRef]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. Garraway, MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef]
- Mehta, P.P.; Kung, P.P.; Yamazaki, S.; Walls, M.; Shen, A.; Nguyen, L.; Gehring, M.R.; Los, G.; Smeal, T.; Yin, M.J. RETRACTED: A novel class of specific Hsp90 small molecule inhibitors demonstrate in vitro and in vivo anti-tumor activity in human melanoma cells. Cancer Lett. 2011, 300, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Marmarelis, M.E.; Hodi, F.S. Activity of the heat shock protein 90 inhibitor ganetespib in melanoma. PLoS ONE 2013, 8, e56134. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Chen, Y.A.; Gibney, G.T.; Weber, J.S.; Kudchadkar, R.R.; Khushalani, N.I.; Markowitz, J.; Brohl, A.S.; Tetteh, L.F.; Ramadan, H.; et al. Combined BRAF and HSP90 Inhibition in Patients with Unresectable BRAF (V600E)-Mutant Melanoma. Clin. Cancer Res. 2018, 24, 5516–5524. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.; Xu, X.; Davies, M.A.; Gimotty, P.A.; Morrissette, J.J.D.; Tetzlaff, M.T.; Wani, K.M.; Liu, S.; Deng, W.; Buckley, M.; et al. A Multicenter Phase I Study Evaluating Dual PI3K and BRAF Inhibition with PX-866 and Vemurafenib in Patients with Advanced BRAF V600-Mutant Solid Tumors. Clin. Cancer Res. 2018, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Bowles, D.W.; Falchook, G.S.; Messersmith, W.A.; George, G.C.; O’Bryant, C.L.; Vo, A.C.; Klucher, K.; Herbst, R.S.; Eckhardt, S.G.; et al. A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 4173–4182. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Salas Fragomeni, R.A.; Schalck, A.; Ferreiro-Neira, I.; Hoff, T.; Cooper, Z.A.; Haq, R.; Panka, D.J.; Kwong, L.N.; Davies, M.A.; et al. Clinical profiling of BCL-2 family members in the setting of BRAF inhibition offers a rationale for targeting de novo resistance using BH3 mimetics. PLoS ONE 2014, 9, e101286. [Google Scholar] [CrossRef] [PubMed]
- Bhatty, M.; Kato, S.; Piha-Paul, S.A.; Naing, A.; Subbiah, V.; Huang, H.J.; Karp, D.D.; Tsimberidou, A.M.; Zinner, R.G.; Hwu, W.J.; et al. Phase 1 study of the combination of vemurafenib, carboplatin, and paclitaxel in patients with BRAF-mutated melanoma and other advanced malignancies. Cancer 2019, 125, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Plimack, E.R.; Desai, J.R.; Issa, J.P.; Jelinek, J.; Sharma, P.; Vence, L.M.; Bassett, R.L.; Ilagan, J.L.; Papadopoulos, N.E.; Hwu, W.J. A phase I study of decitabine with pegylated interferon alpha-2b in advanced melanoma: Impact on DNA methylation and lymphocyte populations. Investig. New Drugs 2014, 32, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Leon-Ferre, R.; Laux, D.; Deutsch, J.; Smith, B.J.; Frees, M.; Milhem, M. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother. Pharmacol. 2014, 74, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Zakharia, Y.; Monga, V.; Swami, U.; Bossler, A.D.; Freesmeier, M.; Frees, M.; Khan, M.; Frydenlund, N.; Srikantha, R.; Vanneste, M.; et al. Targeting epigenetics for treatment of BRAF mutated metastatic melanoma with decitabine in combination with vemurafenib: A phase lb study. Oncotarget 2017, 8, 89182–89193. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, J.C.; Suvada, J.; Dunkel, I.J.; Casanova, M.; Zhang, W.; Ritchie, N.; Choi, Y.; Park, J.; Das Thakur, M.; Simko, S.; et al. BRIM-P: A phase I, open-label, multicenter, dose-escalation study of vemurafenib in pediatric patients with surgically incurable, BRAF mutation-positive melanoma. Pediatr. Blood Cancer 2018, 65, e26947. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hodi, F.S.; Callahan, M.; Konto, C.; Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 2013, 368, 1365–1366. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Lawson, D.H.; Salama, A.K.; Koon, H.B.; Guthrie, T., Jr.; Thomas, S.S.; O’Day, S.J.; Shaheen, M.F.; Zhang, B.; Francis, S.; et al. Phase II study of vemurafenib followed by ipilimumab in patients with previously untreated BRAF-mutated metastatic melanoma. J. Immunother. Cancer 2016, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.I.; Singh, J.; Ernstoff, M.S.; Lao, C.D.; Flaherty, L.E.; Logan, T.F.; Curti, B.; Agarwala, S.S.; Taback, B.; Cranmer, L.; et al. A multi-center phase II study of high dose interleukin-2 sequenced with vemurafenib in patients with BRAF-V600 mutation positive metastatic melanoma. J. Immunother. Cancer 2018, 6, 76. [Google Scholar] [CrossRef]
- Mooradian, M.J.; Reuben, A.; Prieto, P.A.; Hazar-Rethinam, M.; Frederick, D.T.; Nadres, B.; Piris, A.; Juneja, V.; Cooper, Z.A.; Sharpe, A.H.; et al. A phase II study of combined therapy with a BRAF inhibitor (vemurafenib) and interleukin-2 (aldesleukin) in patients with metastatic melanoma. Oncoimmunology 2018, 7, e1423172. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Stephens, R.; Vecchio, M.D.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Giacomo, A.M.D.; et al. KEYNOTE-022 Part 3: Phase 2 Randomized Study of First-Line Dabrafenib and Trametinib Plus Pembrolizumab or Placebo for BRAF-Mutant Advanced Melanoma; European Association for Medical Oncology: Munich, Germany, 2018; pp. viii442–viii466. [Google Scholar]
- Meier, S.; Baumert, B.G.; Maier, T.; Wellis, G.; Burg, G.; Seifert, B.; Dummer, R. Survival and prognostic factors in patients with brain metastases from malignant melanoma. Onkologie 2004, 27, 145–149. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Maio, M.; Arance, A.; Nathan, P.; Blank, C.; Avril, M.F.; Garbe, C.; Hauschild, A.; Schadendorf, D.; Hamid, O.; et al. Vemurafenib in metastatic melanoma patients with brain metastases: An open-label, single-arm, phase 2, multicentre study. Ann. Oncol. 2017, 28, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Saiag, P.; Robert, C.; Grob, J.J.; Flaherty, K.T.; Arance, A.; Chiarion-Sileni, V.; Thomas, L.; Lesimple, T.; Mortier, L.; et al. Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): A multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 863–873. [Google Scholar] [CrossRef]
- Amaria, R.N.; Prieto, P.A.; Tetzlaff, M.T.; Reuben, A.; Andrews, M.C.; Ross, M.I.; Glitza, I.C.; Cormier, J.; Hwu, W.J.; Tawbi, H.A.; et al. Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: A single-centre, open-label, randomised, phase 2 trial. Lancet Oncol. 2018, 19, 181–193. [Google Scholar] [CrossRef]
- Schadendorf, D.; Long, G.V.; Stroiakovski, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion-Sileni, V.; Schachter, J.; Garbe, C.; Dutriaux, C.; et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Kroeze, S.G.; Fritz, C.; Hoyer, M.; Lo, S.S.; Ricardi, U.; Sahgal, A.; Stahel, R.; Stupp, R.; Guckenberger, M. Toxicity of concurrent stereotactic radiotherapy and targeted therapy or immunotherapy: A systematic review. Cancer Treat. Rev. 2017, 53, 25–37. [Google Scholar] [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Tanita, K.; Sato, Y.; Hidaka, T.; Otsuka, A.; Tanaka, H.; Furudate, S.; Hashimoto, A.; Aiba, S. Two cases of Vogt-Koyanagi Harada disease-like uveitis developing from an advanced melanoma patient treated by sequential administration of nivolumab and dabrafenib/ trametinib therapy. J. Dermatol. 2018, 45, 735–737. [Google Scholar] [CrossRef]
- Muto, Y.; Ng, W.; Namikawa, K.; Takahashi, A.; Tsutsumida, A.; Nishida, M.; Yamazaki, N. Success of rechallenging dabrafenib and trametinib combination therapy after trametinib-induced rhabdomyolysis: A case report. Melanoma Res. 2018, 28, 151–154. [Google Scholar] [CrossRef]
- Irimada, M.; Fujimura, T.; Kambayashi, Y.; Tsukada, A.; Takahashi, T.; Hashimoto, A.; Aiba, S. Severe rhabdomyolysis developing from an advanced melanoma patient treated by pembrolizumab followed by dabrafenib trametinib combined therapy. J. Dermatol. 2019, in press. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-Year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]


{kind=link}
| #No. | Retrieval Style | No. of References |
|---|---|---|
| #01 | “Melanoma/ BRAF or MEK” [TIAB] | 19,869 |
| #02 | “Melanoma/ BRAF” [TIAB] or “BRAF inhibitors” [TIAB] | 5929 |
| #03 | “Melanoma/ BRAF inhibitors” [TIAB] and/ or “MEK inhibitors” [TIAB]) | 10,517 |
| #04 | “Melanoma/ BRAF inhibitors” [TIAB] and/ or “MEK inhibitors and mouse” [TIAB] | 4957 |
| #05 | “Melanoma/ BRAF inhibitors” [TIAB] and “mouse” [TIAB] | 471 |
| #06 | “Melanoma/ BRAF inhibitors” [TIAB] and “clinical trials” [TIAB] | 344 |
| #07 | “Melanoma/ BRAF inhibitors” [TIAB] or “MEK inhibitors” [TIAB] and “adverse event” [TIAB] | 2613 |
| #08 | “Melanoma/ BRAF inhibitors” [TIAB] and “adverse events” [TIAB] | 54 |
| #09 | “Melanoma/ BRAF inhibitors” [TIAB] and “clinical trials” [TIAB] and “adverse events” [TIAB] | 17 |
| #10 | “Melanoma” [TIAB] and “immunological background” [TIAB] | 214 |
| #11 | “Melanoma” [TIAB] and “tumor infiltrating leukocytes” [TIAB] | 1793 |
| #12 | “Melanoma” [TIAB] and “immune checkpoint inhibitors” [TIAB] | 1439 |
| #13 | “Melanoma/ BRAF inhibitors” [TIAB] or “MEK inhibitors” [TIAB] and “drug resistance” [TIAB] | 373 |
| Category | Target Molecules | Output | Target Cells | Ref. |
|---|---|---|---|---|
| Drug resistance | aryl hydrocarbon receptor (AhR) | differentiation | melanoma cells | [28] |
| reactive oxygen species (ROS) | induction of apoptosis | drug-resistant tumor cells | [31] | |
| HDAC, pan-CDK | reduce cell viability | drug-resistant tumor cells | [32] | |
| block of proliferation 1 (BOP1) | decrease MAP signaling | drug-resistant in melanoma cells | [29] | |
| Immune regulation | CSF-1 receptor | reduction of myeloid cells | tumor-infiltrating myeloid cells | [22] |
| cell growth | suppress tumor growth | CD8+ T cells | [34] | |
| 4-1BB, OX40 | suppress tumor growth | CD8+ T cells | [35] |
| Protocol | Efficacy (%) | Median OS (Months) | Median PFS (Months) | Ref. |
|---|---|---|---|---|
| vemurafenib monotherapy | 48~52 | 13.6~17.4 | 6.9~7.3 | [2,3,9,36] |
| dabrafenib monotherapy | 50~51 | 13.6 | 5.1 | [2,3,5,10,36] |
| encorafenib monotherapy | 41 | 23.5 | 7.3 | [11] |
| dabrafenib + trametinib combined therapy | 67~83 | 11.4 | [2,3,5,10,36] | |
| vemurafenib + cobimetinib combined therapy | 22.3 | 12.4 | [9] | |
| encorafenib + binimetinib combined therapy | 64 | 33.6 | 19 | [11] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimura, T.; Fujisawa, Y.; Kambayashi, Y.; Aiba, S. Significance of BRAF Kinase Inhibitors for Melanoma Treatment: From Bench to Bedside. Cancers 2019, 11, 1342. https://doi.org/10.3390/cancers11091342
Fujimura T, Fujisawa Y, Kambayashi Y, Aiba S. Significance of BRAF Kinase Inhibitors for Melanoma Treatment: From Bench to Bedside. Cancers. 2019; 11(9):1342. https://doi.org/10.3390/cancers11091342
Chicago/Turabian StyleFujimura, Taku, Yasuhiro Fujisawa, Yumi Kambayashi, and Setsuya Aiba. 2019. "Significance of BRAF Kinase Inhibitors for Melanoma Treatment: From Bench to Bedside" Cancers 11, no. 9: 1342. https://doi.org/10.3390/cancers11091342
APA StyleFujimura, T., Fujisawa, Y., Kambayashi, Y., & Aiba, S. (2019). Significance of BRAF Kinase Inhibitors for Melanoma Treatment: From Bench to Bedside. Cancers, 11(9), 1342. https://doi.org/10.3390/cancers11091342