Mebendazole as a Candidate for Drug Repurposing in Oncology: An Extensive Review of Current Literature

 , , , and
, , , and
Abstract
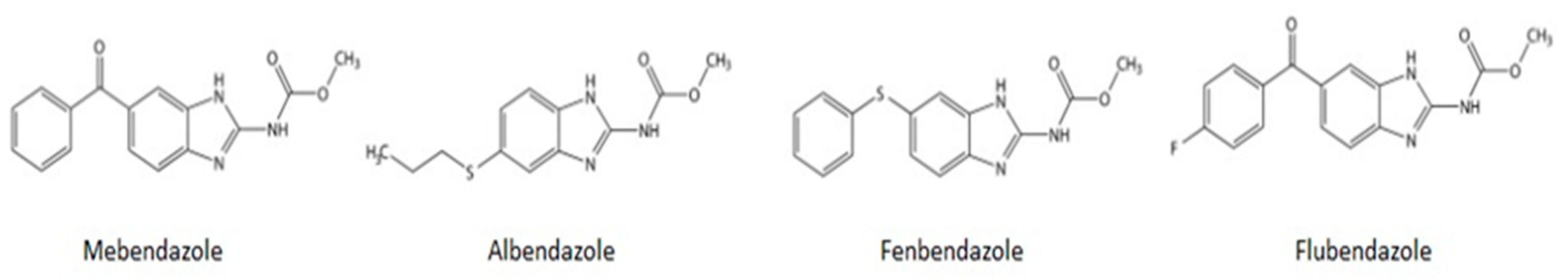
1. Introduction
2. Current Indications and Safety
3. Pharmacokinetics and Pharmacodynamics
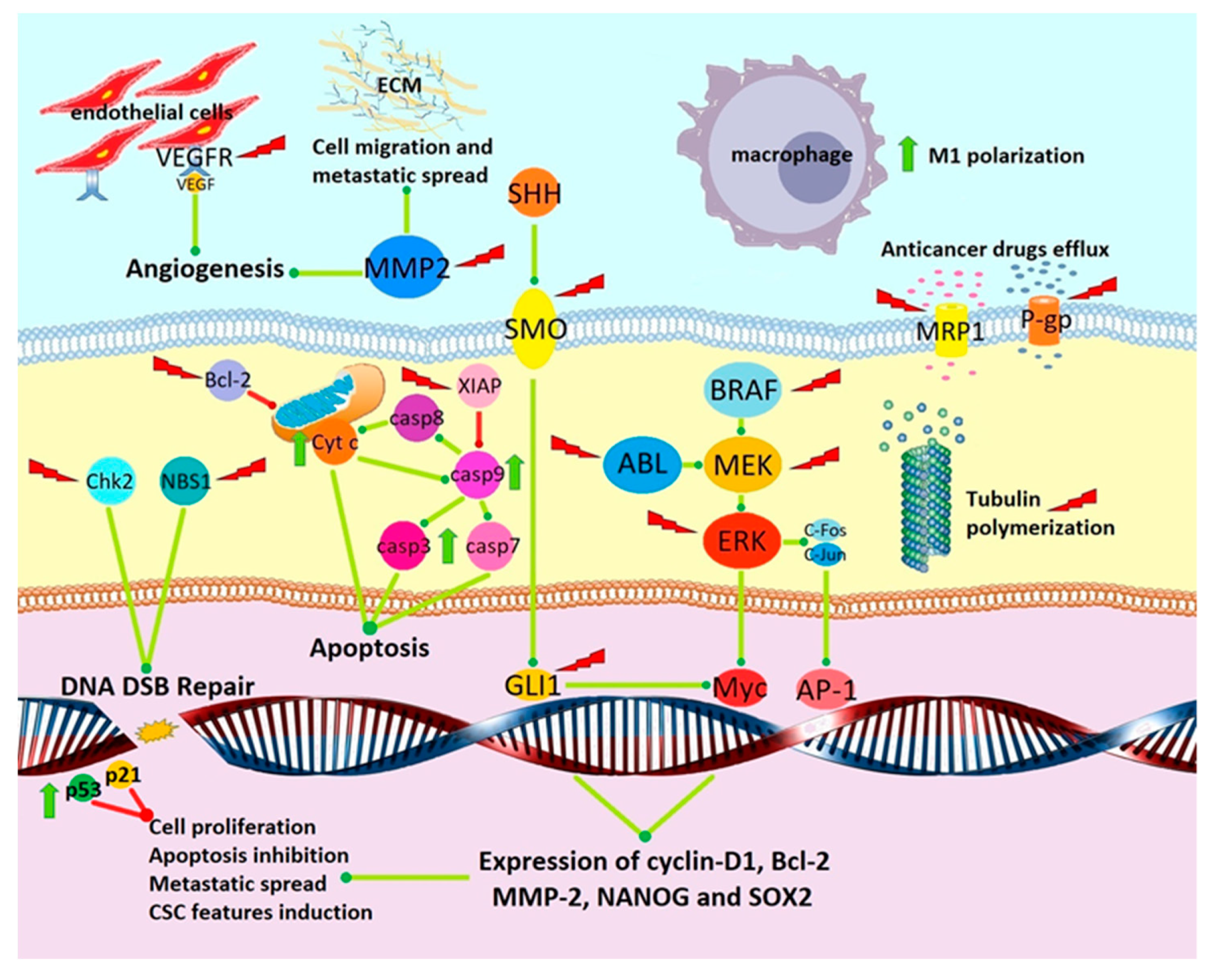
4. Preclinical Evidence of Anticancer Activity
4.1. Tubulin Depolymerization
4.2. Angiogenesis Inhibition
4.3. Inhibition of Signal Transduction Pathways Involved in Cancer Progression
4.4. Sensitization to Chemotherapy and Radiotherapy
4.5. Induction of Apoptosis and Cytotoxicity
4.6. Inhibition of Kinases
4.7. Induction of Antitumor Immune Response
5. Clinical Evidence of Anticancer Activity
6. Conclusions
Funding
Conflicts of Interest
References
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar] [PubMed]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed]
- Wein, L.; Loi, S. Mechanisms of resistance of chemotherapy in early-stage triple negative breast cancer (TNBC). Breast 2017, 34, S27–S30. [Google Scholar] [CrossRef] [PubMed]
- Velaei, K.; Samadi, N.; Barazvan, B.; Rad, J.S. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast 2016, 30, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yeldag, G.; Rice, A.; Del Río Hernández, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; Van Waes, C.; Allen, C.T. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol. 2016, 58, 59–70. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Vidal, S.J.; Rodriguez-Bravo, V.; Galsky, M.; Cordon-Cardo, C.; Domingo-Domenech, J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene 2014, 33, 4451. [Google Scholar] [CrossRef]
- Scannel, J.W. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Laifenfeld, D.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef] [PubMed]
- WHO Informal Working Group on Echinococcosis. Guidelines for treatment of cystic and alveolar echinococcosis in humans. Bull. World Health Organ. 1996, 74, 231–242. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8789923 (accessed on 30 August 2019).
- Todorov, T.; Vutova, K.; Mechkov, G.; Georgiev, P.; Petkov, D.; Tonchev, Z.; Nedelkov, G. Chemotherapy of human cystic echinococcosis: Comparative efficacy of mebendazole and albendazole. Ann. Trop Med. Parasitol. 1992, 86, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Messaritakis, J.; Psychou, P.; Nicolaidou, P.; Karpathios, T.; Syriopoulou, B.; Fretzayas, A.; Krikos, F.; Matsaniotis, N. High mebendazole doses in pulmonary and hepatic hydatid disease. Arch. Dis. Child. 1991, 66, 532–533. [Google Scholar] [CrossRef] [PubMed]
- Riassunto Delle Caratteristiche Del Prodotto. Available online: https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet?pdfFileName=footer_001445_023821_RCP.pdf&retry=0&sys=m0b1l3 (accessed on 30 August 2019).
- Fernández-Bañares, F.; González-Huix, F.; Xiol, X.; Catalá, I.; Miró, J.; López, N.; Casais, L. Marrow aplasia during high dose mebendazole treatment. Am. J. Trop. Med. Hyg. 1986, 35, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Colle, I.; Naegels, S.; Hoorens, A.; Hautekeete, M. Granulomatous hepatitis due to mebendazole. J. Clin. Gastroenterol. 1999, 28, 44–45. [Google Scholar] [CrossRef]
- Dawson, M.; Braithwaite, P.A.; Roberts, M.S.; Watson, T.R. The pharmacokinetics and bioavailability of a tracer dose of [3H]-mebendazole in man. Br. J. Clin. Pharmacol. 1985, 19, 79–86. [Google Scholar] [CrossRef]
- Dawson, M.; Allan, R.J.; Watson, T.R. The pharmacokinetics and bioavailability of mebendazole in man: A pilot study using [3H]-mebendazole. Br. J. Clin. Pharmacol. 1982, 14, 453–455. [Google Scholar] [CrossRef]
- Braithwaite, P.A.; Roberts, M.S.; Allan, R.J.; Watson, T.R. Clinical pharmacokinetics of high dose mebendazole in patients treated for cystic hydatid disease. Eur. J. Clin. Pharmacol. 1982, 22, 161–169. [Google Scholar] [CrossRef]
- Bekhti, A. Serum concentrations of mebendazole in patients with hydatid disease. Int. J. Clin. Pharmacol. Ther. Toxicol. 1985, 23, 633–641. [Google Scholar] [PubMed]
- Corti, N.; Heck, A.; Rentsch, K.; Zingg, W.; Jetter, A.; Stieger, B.; Pauli-Magnus, C. Effect of ritonavir on the pharmacokinetics of the benzimidazoles albendazole and mebendazole: An interaction study in healthy volunteers. Eur. J. Clin. Pharmacol. 2009, 65, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Münst, G.J.; Karlaganis, G.; Bircher, J. Plasma concentrations of mebendazole during treatment of echinococcosis: Preliminary results. Eur. J. Clin. Pharmacol. 1980, 17, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Dayan, A.D. Albendazole, mebendazole and praziquantel. Review of non-clinical toxicity and pharmacokinetics. Acta. Trop. 2003, 86, 141–159. [Google Scholar] [CrossRef]
- Chico, L.K.; Van Eldik, L.J.; Watterson, D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 2009, 8, 892–909. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.Y.; Staedtke, V.; Wanjiku, T.; Rudek, M.A.; Joshi, A.; Gallia, G.L.; Riggins, G.J. Brain Penetration and Efficacy of Different Mebendazole Polymorphs in a Mouse Brain Tumor Model. Clin. Cancer Res. 2015, 21, 3462–3470. [Google Scholar] [CrossRef] [PubMed]
- Jornet, D.; Bosca, F.; Andreu, J.M.; Domingo, L.R.; Tormos, R.; Miranda, M.A. Analysis of mebendazole binding to its target biomolecule by laser flash photolysis. J. Photochem. Photobiol. B 2016, 155, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, J.; Ramesh, R.; Chada, S.; Gomyo, Y.; Roth, J.A.; Mukhopadhyay, T. The anthelmintic drug mebendazole induces mitotic arrest and apoptosis by depolymerizing tubulin in non-small cell lung cancer cells. Mol. Cancer Ther. 2002, 1, 1201–1209. [Google Scholar]
- Bai, R.Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro. Oncol. 2011, 13, 974–982. [Google Scholar] [CrossRef]
- De Witt, M.; Gamble, A.; Hanson, D.; Markowitz, D.; Powell, C.; Symons, M.; Atlas, M.; Boockvar, J.; Ruggieri, R.; Symons, M. Repurposing Mebendazole as a Replacement for Vincristine for the treatment of Brain tumors. Mol. Med. 2017, 23, 50–56. [Google Scholar] [CrossRef]
- Pinto, L.C.; Soares, B.M.; Pinheiro, J.D.; Riggins, G.J.; Assumpção, P.P.; Burbano, R.M.; Montenegro, R.C. The anthelmintic drug mebendazole inhibits growth, migration and invasion in gastric cancer cell model. Toxicol. In Vitro 2015, 29, 2038–2044. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, T.; Sasaki, J.; Ramesh, R.; Roth, J.A. Mebendazole elicits a potent antitumor effect on human cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 2002, 8, 2963–2969. [Google Scholar] [PubMed]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer. 2012, 12, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.Y.; Staedtke, V.; Rudin, C.M.; Bunz, F.; Riggins, G.J. Effective treatment of diverse medulloblastoma models with mebendazole and its impact on tumor angiogenesis. Neuro. Oncol. 2015, 17, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Williamson, T.; Bai, R.Y.; Staedtke, V.; Huso, D.; Riggins, G.J. Mebendazole and a non-steroidal anti-inflammatory combine to reduce tumor initiation in a colon cancer preclinical model. Oncotarget 2016, 7, 68571–68584. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.J.; Kim, H.K.; Hong, Y.K.; Joe, Y.A. Autophagy Is a Potential Target for Enhancing the Anti-Angiogenic Effect of Mebendazole in Endothelial Cells. Biomol. Ther. 2019, 27, 117–125. [Google Scholar] [CrossRef]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef]
- Larsen, A.R.; Bai, R.Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.M.; Riggins, G.J.; Bunz, F. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Mol. Cancer Ther. 2015, 14, 3–13. [Google Scholar] [CrossRef]
- Hiscutt, E.L.; Hill, D.S.; Martin, S.; Kerr, R.; Harbottle, A.; Lovat, P.E.; Redfern, C.P.; Fulda, S.; Armstrong, J.L.; Lovat, P.E. Targeting X-linked inhibitor of apoptosis protein to increase the efficacy of endoplasmic reticulum stress-induced apoptosis for melanoma therapy. J. Investig. Dermatol. 2010, 130, 2250–2258. [Google Scholar] [CrossRef]
- Doudican, N.A.; Byron, S.A.; Pollock, P.M.; Orlow, S.J. XIAP downregulation accompanies mebendazole growth inhibition in melanoma xenografts. Anticancer Drugs 2013, 24, 181–188. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Simbulan-Rosenthal, C.M.; Dakshanamurthy, S.; Gaur, A.; Chen, Y.S.; Fang, H.B.; Abdussamad, M.; Zhou, H.; Zapas, J.; Calvert, V.; Petricoin, E.F.; et al. The repurposed anthelmintic mebendazole in combination with trametinib suppresses refractory NRASQ61K melanoma. Oncotarget 2017, 8, 12576–12595. [Google Scholar] [CrossRef] [PubMed]
- Fry, E.A.; Inoue, K. C-MYB and DMTF1 in Cancer. Cancer Investig. 2019, 37, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Walf-Vorderwülbecke, V.; Pearce, K.; Brooks, T.; Hubank, M.; van den Heuvel-Eibrink, M.M.; Zwaan, C.M.; Adams, S.; Edwards, D.; Bartram, J.; Samarasinghe, S.; et al. Targeting acute myeloid leukemia by drug-induced c-MYB degradation. Leukemia. 2018, 32, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Joyce, H.; McCann, A.; Clynes, M.; Larkin, A. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin. Drug Metab. Toxicol. 2015, 11, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Celestino Pinto, L.; de Fátima Aquino Moreira-Nunes, C.; Soares, B.M.; Burbano, R.M.R.; de Lemos, J.A.R.; Montenegro, R.C. Mebendazole, an antiparasitic drug, inhibits drug transporters expression in preclinical model of gastric peritoneal carcinomatosis. Toxicol. In Vitro 2017, 43, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Kralova, V.; Hanušová, V.; Caltová, K.; Špaček, P.; Hochmalová, M.; Skálová, L.; Rudolf, E. Flubendazole and mebendazole impair migration and epithelial to mesenchymal transition in oral cell lines. Chem. Biol. Interact. 2018, 293, 124–132. [Google Scholar]
- Poruchynsky, M.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Kumar, M.T.; Robey, R.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar]
- Markowitz, D.; Ha, G.; Ruggieri, R.; Symons, M. Microtubule-targeting agents can sensitize cancer cells to ionizing radiation by an interphase-based mechanism. Onco. Targets Ther. 2017, 10, 5633–5642. [Google Scholar] [CrossRef]
- Zhang, L.; Bochkur, D.M.; Yazal, T.; Dong, K.; Nguyen, A.; Yu, G.; Dao, A.; Bochkur, D.M.; Duhachek-Muggy, S.; Bhat, K.; et al. Mebendazole Potentiates Radiation Therapy in Triple-Negative Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 195–207. [Google Scholar] [CrossRef]
- Coyne, C.P.; Jones, T.; Bear, R. Anti-Neoplastic Cytotoxicity of Gemcitabine-(C4-amide)-[anti-EGFR] in Dual-combination with Epirubicin-(C3-amide)-[anti-HER2/neu] against Chemotherapeutic-Resistant Mammary Adenocarcinoma (SKBr-3) and the Complementary Effect of Mebendazole. J. Cancer Res. Ther. Oncol. 2014, 2, 203. [Google Scholar] [CrossRef] [PubMed]
- Kipper, F.C.; Silva, A.O.; Marc, A.L.; Confortin, G.; Junqueira, A.V.; Neto, E.P.; Lenz, G. Vinblastine and antihelmintic mebendazole potentiate temozolomide in resistant gliomas. Investig. New Drugs. 2018, 36, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Buttrick, S.; Shah, A.H.; Komotar, R.J.; Ivan, M.E. Management of Atypical and Anaplastic Meningiomas. Neurosurg. Clin. N. Am. 2016, 27, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, C.G.; Williamson, T.; Riggins, G.J. Mebendazole and radiation in combination increase survival through anticancer mechanisms in an intracranial rodent model of malignant meningioma. J. Neurooncol. 2018, 140, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, Y.; Zhang, H.; Huang, E.; Gao, L.; Luo, W.; Wei, Q.; Fan, J.; Song, D.; Liao, J.; et al. Anthelmintic Mebendazole enhances cisplatin’s effect on suppressing cell proliferation and promotes differentiation of head and neck squamous cell carcinoma (HNSCC). Oncotarget 2017, 8, 12968–12982. [Google Scholar] [PubMed]
- Martarelli, D.; Pompei, P.; Baldi, C.; Mazzoni, G. Mebendazole inhibits growth of human adrenocortical carcinoma cell lines implanted in nude mice. Cancer Chemother. Pharmacol. 2008, 61, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Doudican, N.; Rodriguez, A.; Osman, I.; Orlow, S.J. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol. Cancer Res. 2008, 6, 1308–1315. [Google Scholar] [CrossRef]
- Pinto, L.C.; Mesquita, F.P.; Soares, B.M.; da Silva, E.L.; Puty, B.; de Oliveira, E.H.; Burbano, R.R.; Montenegro, R.C. Mebendazole induces apoptosis via C-MYC inactivation in malignant ascites cell line (AGP01). Toxicol. In Vitro 2019, 60, 305–312. [Google Scholar] [CrossRef]
- Dakshanamurthy, S.; Issa, N.T.; Assefnia, S.; Seshasayee, A.; Peters, O.J.; Madhavan, S.; Uren, A.; Brown, M.L.; Byers, S.W. Predicting New Indications for Approved Drugs Using a Proteo-Chemometric Method. J. Med. Chem. 2012, 55, 6832–6848. [Google Scholar] [CrossRef]
- Issa, N.T.; Peters, O.J.; Byers, S.W.; Dakshanamurthy, S. Repurpose VS: A Drug Repurposing-Focused Computational Method for Accurate Drug-Target Signature Predictions. Comb. Chem. High. Throughput Screen 2015, 18, 784–794. [Google Scholar] [CrossRef]
- Yamada, T.; Masuda, M. Emergence of TNIK inhibitors in cancer therapeutics. Cancer Sci. 2017, 108, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Chen, L.; Zhang, S. Comprehensive Modeling and Discovery of Mebendazole as a Novel TRAF2- and NCK interacting Kinase Inhibitor. Sci. Rep. 2016, 6, 33534. [Google Scholar] [CrossRef] [PubMed]
- Nygren, P.; Fryknäs, M.; Agerup, B.; Larsson, R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Blom, K.; Senkowski, W.; Jarvius, M.; Berglund, M.; Rubin, J.; Larsson, R. The anticancer effect of mebendazole may be due to M1 monocyte/macrophage activation via ERK1/2 and TLR8-dependent inflammasome activation. Immunopharmacol. Immunotoxicol. 2017, 39, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.; Mansoori, S.; Blom, K.; Berglund, M.; Lenhammar, L.; Andersson, C.; Loskog, A.; Fryknäs, M.; Nygren, P.; Larsson, R. Mebendazole stimulates CD14+ myeloid cells to enhance T-cell activation and tumour cell killing. Oncotarget 2018, 9, 30805–30813. [Google Scholar] [CrossRef] [PubMed]
- Blom, K.; Rubin, J.; Berglund, M.; Jarvius, M.; Lenhammar, L.; Parrow, V.; Andersson, C.; Loskog, A.; Fryknäs, M.; Nygren, P.; et al. Mebendazole-induced M1 polarization of THP-1 macrophages may involve DYRK1B inhibition. BMC Res. Notes 2019, 12, 234. [Google Scholar] [CrossRef] [PubMed]
- Dobrosotskaya, I.Y.; Hammer, G.D.; Schteingart, D.E.; Maturen, K.E.; Worden, F.P. Mebendazole monotherapy and long-term disease control in metastatic adrenocortical carcinoma. Endocr. Pract. 2011, 17, e59–e62. [Google Scholar] [CrossRef]
- Nygren, P.; Larsson, R. Drug repositioning from bench to bedside: Tumour remission by the antihelmintic drug mebendazole in refractory metastatic colon cancer. Acta. Oncol. 2014, 53, 427–428. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)—Mebendazole as an anti-cancer agent. Ecancermedicalscience 2014, 8, 443. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, S.; Nishimura, Y.; Kon, T.; Yamanaka, Y.; Kawase, R.; Tanaka, T. DNA Damage Response Is Involved in the Developmental Toxicity of Mebendazole in Zebrafish Retina. Front. Pharmacol. 2016, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Verduzco, D.; Gatenby, R. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef]
- Lien, K.; Georgsdottir, S.; Sivanathan, L.; Chan, K.; Emmenegger, U. Low-dose metronomic chemotherapy: A systematic literature analysis. Eur. J. Cancer 2013, 49, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P.; Vikas, P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience 2014, 8, 442. [Google Scholar] [CrossRef]
- Arif, S.H.; Shams-Ul-Bari; Wani, N.A.; Zargar, S.A.; Wani, M.A.; Tabassum, R.; Hussain, Z.; Baba, A.A.; Lone, R.A. Albendazole as an adjuvant to the standard surgical management of hydatid cyst liver. Int. J. Surg. 2008, 6, 448–451. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Szwajcer, M.; Chin, M.; Liauw, W.; Seef, J.; Galettis, P.; Morris, D.L.; Links, M. Phase I clinical trial to determine maximum tolerated dose of oral albendazole in patients with advanced cancer. Cancer Chemother. Pharmacol. 2010, 65, 597–605. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Dosage | Cmax and AUC | Half-Life of Elimination | Tissue Concentrations |
|---|---|---|---|
| 10 mg/kg, single dose or chronic administration, 12 patients treated for cystic hydatid disease [21] | Cmax 17.5 to 500 ng/mL (0.06–1.69 µM, mean 0.24 µM) after a single dose. In chronic therapy mean Cmax 0.47 µM and AUC five times higher than after single dose | T1/2 2.8–9.0 h, time to peak plasma concentration 1.5–7.25 h | Concentrations of MBZ found in the tissue and cyst material collected from two patients during surgery ranged from 59.5 to 206.6 ng/g wet weight |
| 1–12 g/day, 17 patients treated for hydatid cysts and 5 volunteers [22] | Cmax 0.03–1.64 µM | T1/2 3.3–11.5 h | - |
| 1000 mg single dose, 8 healthy volunteers [23] | mean Cmax 0.11 µM, mean AUC 207.2 µg·h/L | T1/2 mean 7.4 h | - |
| 1.5 g single dose or repeated 1 g administrations [24] | Cmax 0.017–0.134 µM after single dose and up to 0.5 µM after repeated administrations | - | - |
| Phase | Condition | Intervention | Institution |
|---|---|---|---|
| Phase 1 | Newly diagnosed high-grade glioma | Standard of care (surgery and radio-chemotherapy) followed by MBZ (MTD to define) + adjuvant sequential TMZ. | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins |
| Phase 1 | Pediatric patients affected by medulloblastoma or high-grade glioma in progression after standard therapies | MBZ alone (MTD to define) | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins |
| Phase 1–2 | Pediatric patients affected by low- or high-grade glioma | MBZ 50–200 mg/kg/day (MTD to define) in combination with vincristine, carboplatin, and temozolomide (low grade) or bevacizumab and irinotecan (high-grade glioma) | Cohen Children’s Medical Center of New York |
| Phase 2 | Advanced or metastatic gastrointestinal cancer or cancer of unknown origin | MBZ alone, dose escalation (50–4000 mg), and pharmacokinetic analysis | Uppsala University |
| Phase 2 | Metastatic or advanced cancer (different organs and histology) | MBZ 100 mg b.i.d. + metformin up to 1000 mg b.i.d. + doxycycline 100 mg/die + atorvastatin up to 80 mg/die; “real world setting”, with or without concomitant standard of treatment | Care Oncology Clinic, London |
| Phase 2 | Stage IV colorectal cancer | MBZ (dose not specified) concomitant with adjuvant FOLFOX + bevacizumab | Tanta University |
| Author Year | Cell Line | Ic50 Antiproliferative | Biological Effect |
|---|---|---|---|
| Mukhopadhyay T et al. 2002 [33] | Human Non-Small Cell Lung Cancer (NSCLC): A549, H1299, H460. Human breast, ovary, and colon carcinoma and osteosarcoma | NSCLC cell lines: ~0.16 µM. Other cell lines: 0.1–0.8 μM. | Not specified. Growth inhibition of about five-fold after exposure of H460 and A549 cells to 0.165 μM for 5 days. No effect on HUVECs and normal fibroblasts also at 1 μM. |
| Sasaki J et al. 2002 [29] | Three human NSCLC cell lines | A549 0.417 µM, H1299 0.260 µM, H460 0.203 µM | 0.5 µM tubulin depolymerization. Induction of p53 and p21 expression after 24 h, induction of apoptosis after 48 h in 35% of H460 cells and 15% of A549 cells. |
| Martarelli D et al. 2008 [57] | Human adrenocortical cancer H295R and SW-13 | H295R 0.23 μM, SW-13 0.27 μM | Cell invasion inhibition (0.085 μM). Cytochrome c and caspase-9 and 3 mediated apoptosis. |
| Doudican NA et al. 2008 [41] | Human melanoma M-14 and A-375 | Not specified | Decrease in XIAP levels, increase in apoptosis markers (cleaved PARP and caspase 9) at 0.5 μM. |
| Bai RY et al. 2011 [30] | A panel of 10 glioblastoma cell lines. | Between 0.11 and 0.31 μM | Inhibition of tubulin polymerization in 060919 cells at 0.1 µM for 72 h. |
| Doudican N et al. 2013 [58] | Human melanoma SK-Mel-19 and M-14 | SK-Mel-19 0.32 µM, M14 0.30 µM. | Apoptosis induction at 1 µM for 24 h in 25% of M-14 cells and 31% of SK-Mel-19. At 0.5 µM only 26% of SK-Mel-19 cells maintained proliferative capacity. |
| Nygren P et al. 2013 [64] | Human colon cancer cell lines HT29, HCT-8 and SW626, HCT 116 and RKO | Less than 5 μM for all the lines tested and <1 μM for 3 lines | Inhibition of several kinases (including BCR–ABL and BRAF) in the nanomolar range. |
| Coyne CP et al. 2014 [52] | Human mammary adenocarcinoma SKBr-3 | About 0.35 μM at 96h and 0.25μM at 182 h. IC80 at 182 h ~0.30 μM | Survival fraction reduced to 36.9–9.2% after exposure to 0.2–2.5 μM for 96–182 h. Synergy with anti-HER2 conjugates with anthracyclines or gemcitabine: 0.15 μM MBZ ↓ survival fraction from 48.7% to 7.7% at chemotherapeutic-equivalent concentrations of 10−8 M and from 79.5% to 8.7% at 10−10 M. |
| Larsen AR et al. 2015 [39] | DAOY human medulloblastoma | Not specified | Sonic hedgehog (SHH) pathway inhibition: inhibition o.d. SMO mutant proteins and reduction in GLI1 expression (0.1–1 μM, IC50 0.516 μM). Inhibition of cell proliferation (0.1 μM) and primary cilium assembly, induction of apoptosis (1 μM). |
| Bai RY et al. 2015 [35] | A panel of 8 medulloblastoma cell lines | Between 0.13 and 1 μM after 72 h | Inhibition of VEGFR2 autophosphorylation, at 1–10 μM in cultured HUVECs and with an IC50 of 4.3 μM in a cell-free kinase assay. |
| Pinto LC et al. 2015 [32] | Human gastric cancer ACP-02, ACP-03 and AGP-01 (malignant ascites) | ACP-02 0.39 μM, AGP-01 0.59 μM, ACP-03 1.25 µM | Disruption of microtubules, inhibition of invasion and migration and of MMP-2 activity. |
| Williamson T et al. 2016 [36] | Colo-rectal carcinoma cell lines DLD-1, HCT-116, HT29, and SW480 | DLD-1 0.28 μM, HCT-116 0.25 μM, HT29 0.20 μM, and SW480 0.81 μM | Not specified. |
| Simbulan-Rosenthal CM et al. 2017 [43] | Patient-derived melanoma NRAS mutated (BAK and BUL) and BRAF mutated (STU) | Not specified | Inhibition of several kinases, including BRAF wild type and BRAFV600E (with a Kd of 210 and 230 nM) and MEK. Inhibition of MAPK/ERK pathway, induction of apoptosis, synergy with trametinib |
| Zhang F et al. 2017 [56] | Human head and neck squamous cell carcinoma CAL27 and SCC15 | CAL27 1.28 and SCC15 2.64 μM | Apoptosis induction as a single drug. Strong synergistic effect with cisplatin. Increase in CAL27 and inhibition in SCC15 cells of proliferation related pathways |
| De Witt M et al. 2017 [31] | GL261 murine glioma | Cell viability suppression 160 nM | EC50 for microtubule depolymerization 132 nM, mitotic arrest induction 192 nM |
| Pinto LC et al. 2017 [47] | AGP-01 intestinal type adenocarcinoma | Not specified | Inhibition of P-gp and MRP1 at 1.0 μM for 24 h. Inhibition of MATE1 at 0.1–1.0 μM |
| Blom K et al. 2017 [66] | THP-1 monocyte and HT29 colon cancer co-culture | Not specified | 1–10 μM for 6 h increased release of pro-inflammatory M1 cytokines (such as IL-1β, TNF, IL8, and IL6) and surface markers (CD80 and CD 86), induction of antitumor response in co-culture. Induction of IL-1β secretion in presence (1 μM) or in absence (10 μM) of LPS. Induction of tumor suppressive effect in co-cultures |
| Markowitz D et al. 2017 [50] | Human GBM14 glioblastoma Murine GL261 glioma | Not specified | Radiosensitization with an EC50 of 35 nM. Cytoplasmic sequestration of DDRp Chk2 (EC50 31 nM) and Nbs1 (EC50 25 nM) |
| Walf-Vorderwülbecke V et al. 2018 [45] | Eight different Acute Myeloid Leukemia cell lines | IC50s for cell viability between 0.07 and 0.26 µM | Degradation of c-MYB and inhibition of its expression. Reduction in colony formation (>80% after exposure of THP1 AML cells for 16 h at 10 µM) |
| Rubin J et al. 2018 [67] | Co-culture of PBMCs, A549 cells and human fibroblasts or HUVEC cells | Not specified | 0.3–10 μM increased release of pro-inflammatory cytokines, reduced levels of VEGF and VCAM-1, potentiated killing of A549 NSCLC cells mediated by CD3/IL2 activated PBMCs |
| Skibinski CG et al. 2018 [55] | Seven meningioma cell lines | IC50s for cell viability after 72 h 0.26–0.42 μM | Reduced clonogenic activity, induced cytotoxicity, increased levels of cleaved caspase-3 and PARP and reduced colony formation |
| Kralova V et al. 2018 [48] | PE/CA-PJ15 and H376 oral SCC; DOK premalignant oral keratinocytes | Not specified | PE/CA-PJ15 and H376: 0.1–0.25 μM MBZ or FBZ inhibition of kinases (FAK) and GTPases (Rho-A, Rac1); dose dependent migration inhibition (0.1–5 μM) DOK: TGF-β induced N-cadherin inhibited at 0.05–0.2 μM for 48 h |
| Zhang L et al. 2019 [51] | SUM159PT and MDA-MB-231 TNBC | 0.35 µM in monolayers and 0.4 µM in mammospheres after 72 h. | 0.5 µM arrest in the G2/M phase; significant radiosensitizing effect at all radiation doses tested (1–8 Gy). Mebendazole at 0.35 and 0.7 µM dose-dependent decrease of ALDH1 positive CSCs; Hedgehog pathway inhibition. ↑ fraction of apoptotic cells, ↑ DNA DSBs |
| Sung SJ et al. 2019 [37] | HUVECs | IC50s for cell proliferation after 48 h 0.7–2.5 μM | Inhibition of VEGF or bFGF induced migration (IC50 0.7–0.9 μM) and tube formation (IC50 0.8–1.5 μM); ↑ p53 level up to 2.9 fold Dose and time-dependent apoptosis in up to 34% of cells at 72 h |
| Blom K et al. 2019 [68] | THP-1 monocytes and macrophages. | Not specified | DYRK1B inhibition IC50 of 360 nM and kD of 7 nM. 10 µM for 24 h ↑ M1 marker CD80 and ↓ M2 marker CD163 |
| Pinto LC et al. 2019 [59] | AGP01 gastric cancer | Not specified | 0.5–1 μM ↑ caspase 3 and 7 activity, ↓ C-MYC mRNA and C-MY. Cell cycle arrest in G0/G1 and G2/M phases at 0.5 μM and 1.0 μM. Apoptosis induction 68% (0.5 μM) and 74% (1 μM) of cells at 72 h |
| Author and Year | CELL LINES TESTED | DOSE | BIOLOGICAL EFFECT IN VIVO | ANTITITUMOR EFFECT |
|---|---|---|---|---|
| Mukhopadhyay T et al. 2002 [33] | H460 and A549 human NSCLC. K1735 murine melanoma. | 0.4–0.8–1 mg/mouse/e.o.d. (oral) | Angiogenesis inhibition Metastatic spread inhibition | H460: tumor growth inhibition of 30% (0.4 mg) and 80% (0.8 mg) and almost complete arrest of growth (1 mg/mice/e.o.d.) A549: 80% reduction of metastases number in lungs (1 mg/mouse/e.o.d.) K1735 allograft: 1 mg growth inhibition of ~70%. |
| Martarelli D et al. 2008 [57] | H295R and SW-13 human adrenocortical cancer | 1 or 2 mg/mice/day (oral) | Apoptosis induction Invasion inhibition Metastatic spread inhibition | H295R: about 50% (1 mg) and 60% (2 mg) tumor volume reduction SW-13: about 70% (1 mg) and 60% (2 mg) tumor volume reduction and 50% (1 mg) and 75% (2 mg) reduction of lung metastases number |
| Bai RY et al. 2011 [30] | GL261 murine glioma and 060,919 human GBM | 50 mg/kg (oral) | Not specified | Survival increase in GL261: 29 d CTRL vs. 41 d TMZ vs. 49 d MBZ vs. 50 d TMZ + MBZ vs 36 d ABZ 50 mg/kg vs 39 d ABZ 150 mg/kg Survival increase in 060919 xenograft: 48 d CTRL versus 65 d MBZ vs 43 d ABZ 150 mg/kg |
| Doudican NA et al. 2013 [41] | M-14 human melanoma | 1 or 2 mg/mouse/day (oral by gavage) | XIAP inhibition Apoptosis induction | Tumor growth inhibition of 83% (1 mg) and 77% (2 mg) |
| Larsen AR et al. 2015 [39] | DAOY human medulloblastoma | 25–50 mg/kg (oral) | Sonic Hedgehog pathway inhibition | Survival increase: 75 d control group (CTRL) versus 94 d MBZ 25 mg/kg versus 113 d MBZ 50 mg/kg |
| Bai RY et al. 2015 [35] | D425 human medulloblastoma. Murine parental or SMO-D477G mutated medulloblastoma. | 50 mg/kg/day (oral in food) | Angiogenesis inhibition | Survival increase in murine medulloblastoma: 150% increase in the parental line and 100% in SMO-D477G mutated allograft; growth inhibition in both models. Survival increase in D425 xenograft: 125% increase in survival versus CTRL; tumor burden reduction. |
| Bai RY et al. 2015 [27] | GL261 murine glioma D425 human medulloblastoma | 50 mg/kg of polymorph A, B or C MBZ (oral by gavage) | Not specified | Survival increase, enhanced by elacridar (ELD) GL261:29 d CTRL vs. 34 d ELD vs. 53 d MBZ vs. 92.5 d MBZ + ELD (for 7 days) vs. 110.5 d MBZ + ELD (for 14 days) D425: 24 d CTRL vs. 33 d ELD vs. 52 d MBZ vs. 77 d MBZ + ELD (for 7 days) |
| Williamson T et al. 2016 [36] | HT29 or SW480 human colorectal cancer APCmin/+ model | 50 mg/kg or 35 mg/kg (oral by gavage) | Inhibition of several pathways (MYC, COX2 and Bcl-2) and cytokines. Angiogenesis inhibition. | Tumor volume and weight reduction: respectively 62% and 65% in HT29 and 67% and 59% in SW480 (50 mg/kg) APCmin/+ chemoprevention model: reduction of tumor numbers 56% as a single agent (35 mg/kg) and up to 90% in combination with sulindac |
| Simbulan-Rosenthal CM et al. 2017 [43] | BAK human melanoma | 40 mg/kg (oral by gavage) | MEK1/2 and ERK1/2 inhibition | MBZ or trametinib (1 or 3 mg/kg) showed no growth inhibition as single agents, in combination 50% volume reduction and increased survival |
| Zhang et al. 2017 [56] | CAL27 human head and neck squamous cell carcinoma | 7.5 mg/kg i.p. e.o.d | Cell differentiation | Slight volume increased, induction of cell differentiation (extensive keratinization, diminished expression of proliferation markers and up-regulated expression of differentiation markers). |
| De Witt M et al. 2017 [31] | GL261 murine glioma | 50–100 mg/kg of polymorph C MBZ (oral) | Not specified | Survival increase: 10 d CTRL vs 11 d voncristine vs 17 d MBZ 50 mg/kg vs. 19 d MBZ 100 mg/kg |
| Walf-Vorderwülbecke V et al. 2018 [45] | THP1 human acute myeloid leukemia | 200 mg/kg of diet (oral, mixed in food) | c-MYB degradation | Growth inhibition and survival increase (~ 65 days vs. ~40 days in CTRL group) |
| Skibinski CG et al. 2018 [55] | KT21MG1 human meningioma | 50 mg/kg/day in high fat diet | Apoptosis induction, angiogenesis inhibition | KT21MG1 intercranial xenograft: median survival 19 d in CTRL group, 30 d MBZ 33.5 d RT (12 Gy) and 39 d RT + MBZ |
| Zhang L et al. 2019 [51] | SUM159PT human TNBC | 10 or 20 mg/kg 5 days/week i.p. | Radiosensitization | MBZ alone modest effect, IR 10 Gy evident growth delay potentiated by MBZ 20 mg/kg |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a Candidate for Drug Repurposing in Oncology: An Extensive Review of Current Literature. Cancers 2019, 11, 1284. https://doi.org/10.3390/cancers11091284
Guerini AE, Triggiani L, Maddalo M, Bonù ML, Frassine F, Baiguini A, Alghisi A, Tomasini D, Borghetti P, Pasinetti N, et al. Mebendazole as a Candidate for Drug Repurposing in Oncology: An Extensive Review of Current Literature. Cancers. 2019; 11(9):1284. https://doi.org/10.3390/cancers11091284
Chicago/Turabian StyleGuerini, Andrea Emanuele, Luca Triggiani, Marta Maddalo, Marco Lorenzo Bonù, Francesco Frassine, Anna Baiguini, Alessandro Alghisi, Davide Tomasini, Paolo Borghetti, Nadia Pasinetti, and et al. 2019. "Mebendazole as a Candidate for Drug Repurposing in Oncology: An Extensive Review of Current Literature" Cancers 11, no. 9: 1284. https://doi.org/10.3390/cancers11091284
APA StyleGuerini, A. E., Triggiani, L., Maddalo, M., Bonù, M. L., Frassine, F., Baiguini, A., Alghisi, A., Tomasini, D., Borghetti, P., Pasinetti, N., Bresciani, R., Magrini, S. M., & Buglione, M. (2019). Mebendazole as a Candidate for Drug Repurposing in Oncology: An Extensive Review of Current Literature. Cancers, 11(9), 1284. https://doi.org/10.3390/cancers11091284