Novel Carbazole-Piperazine Hybrid Small Molecule Induces Apoptosis by Targeting BCL-2 and Inhibits Tumor Progression in Lung Adenocarcinoma In Vitro and Xenograft Mice Model

 and
and
Abstract
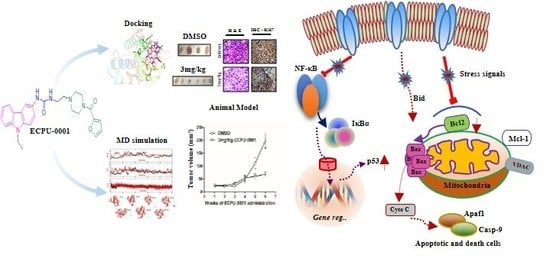
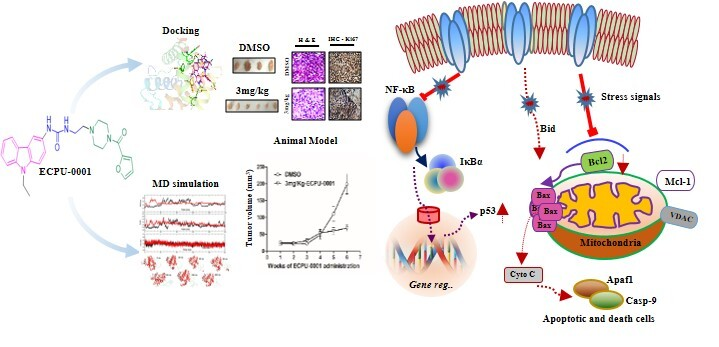{kind=link}
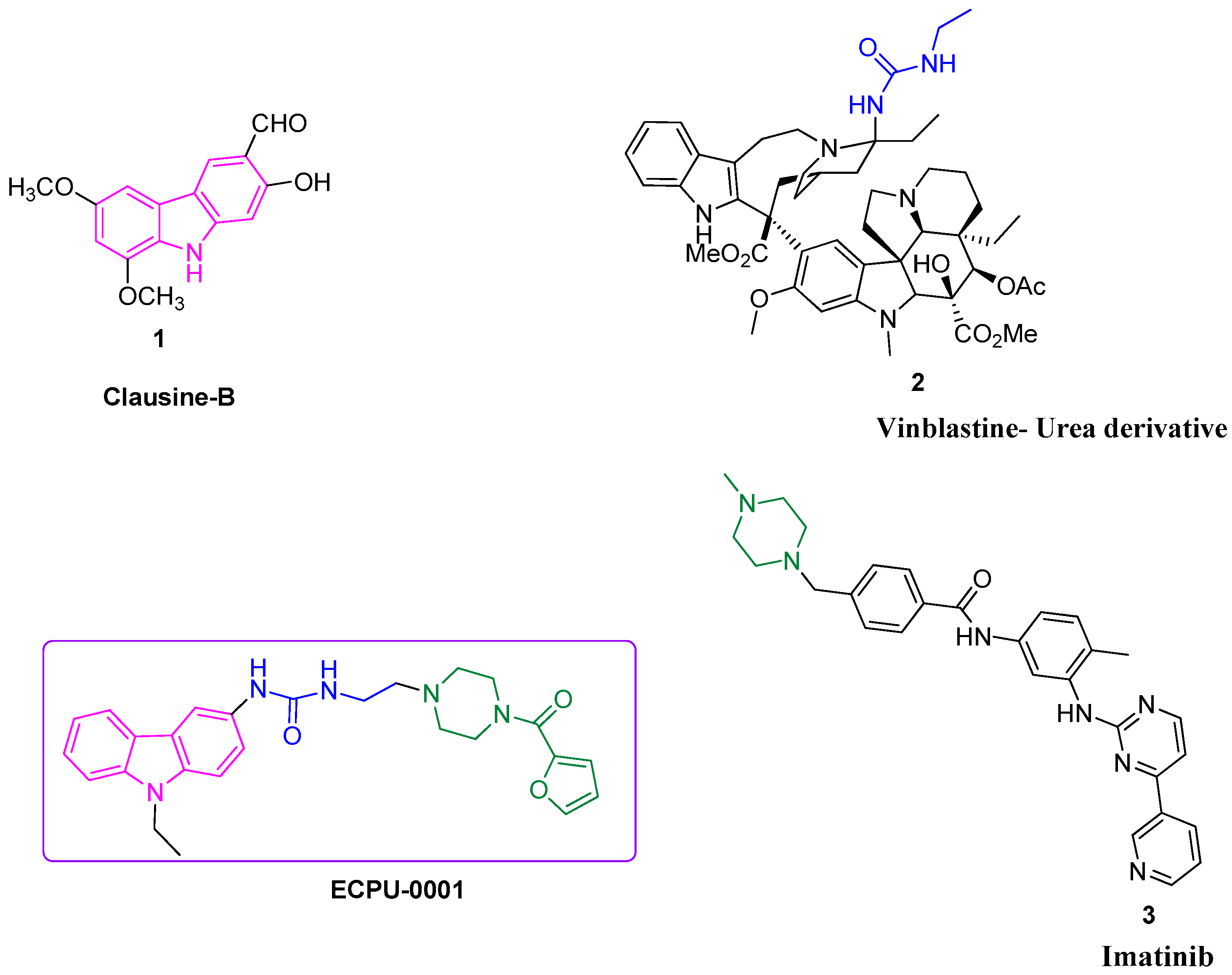{kind=link}
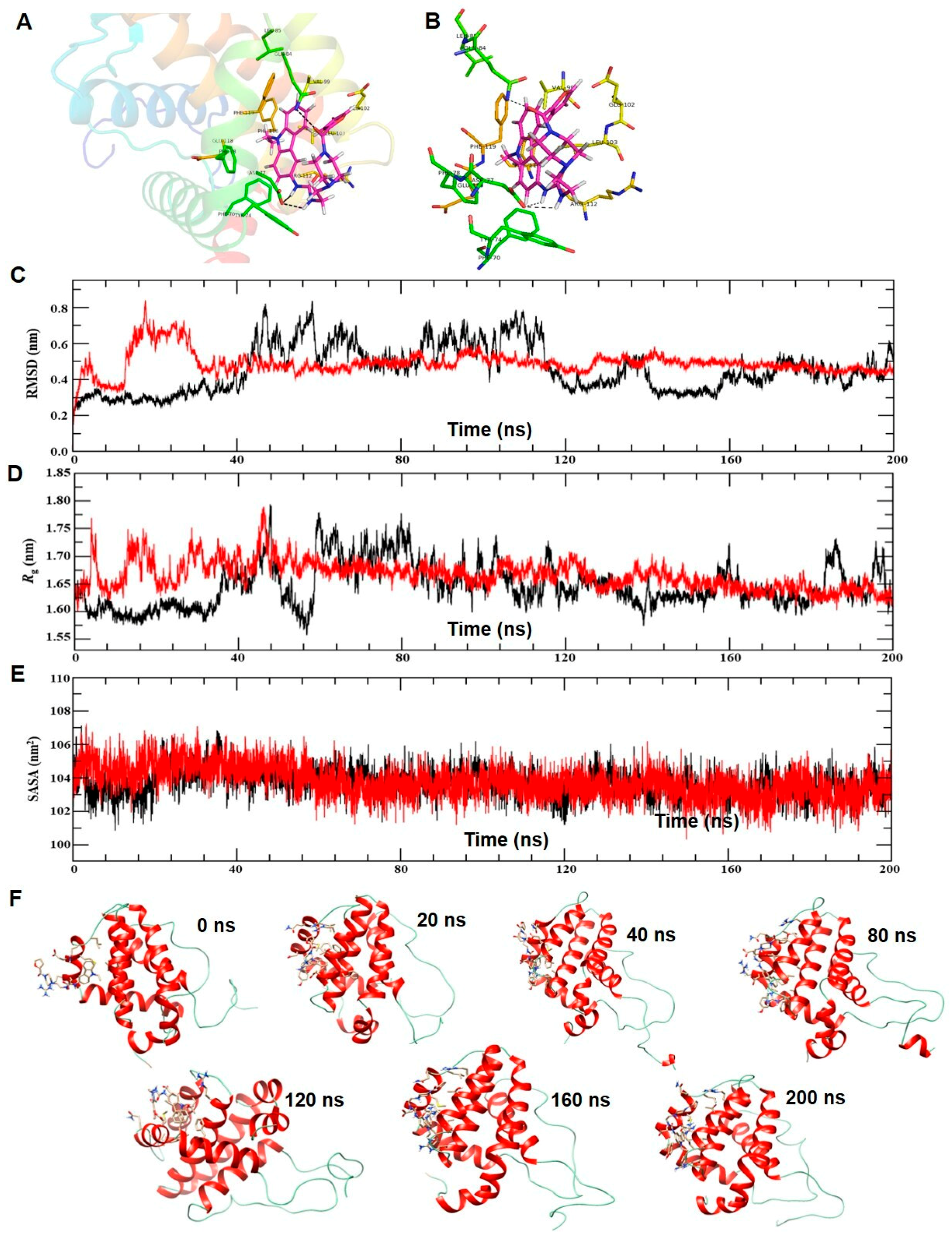{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Molecular Interaction of ECPU-0001 with BCL-2: Molecular Docking and MD Simulation Study
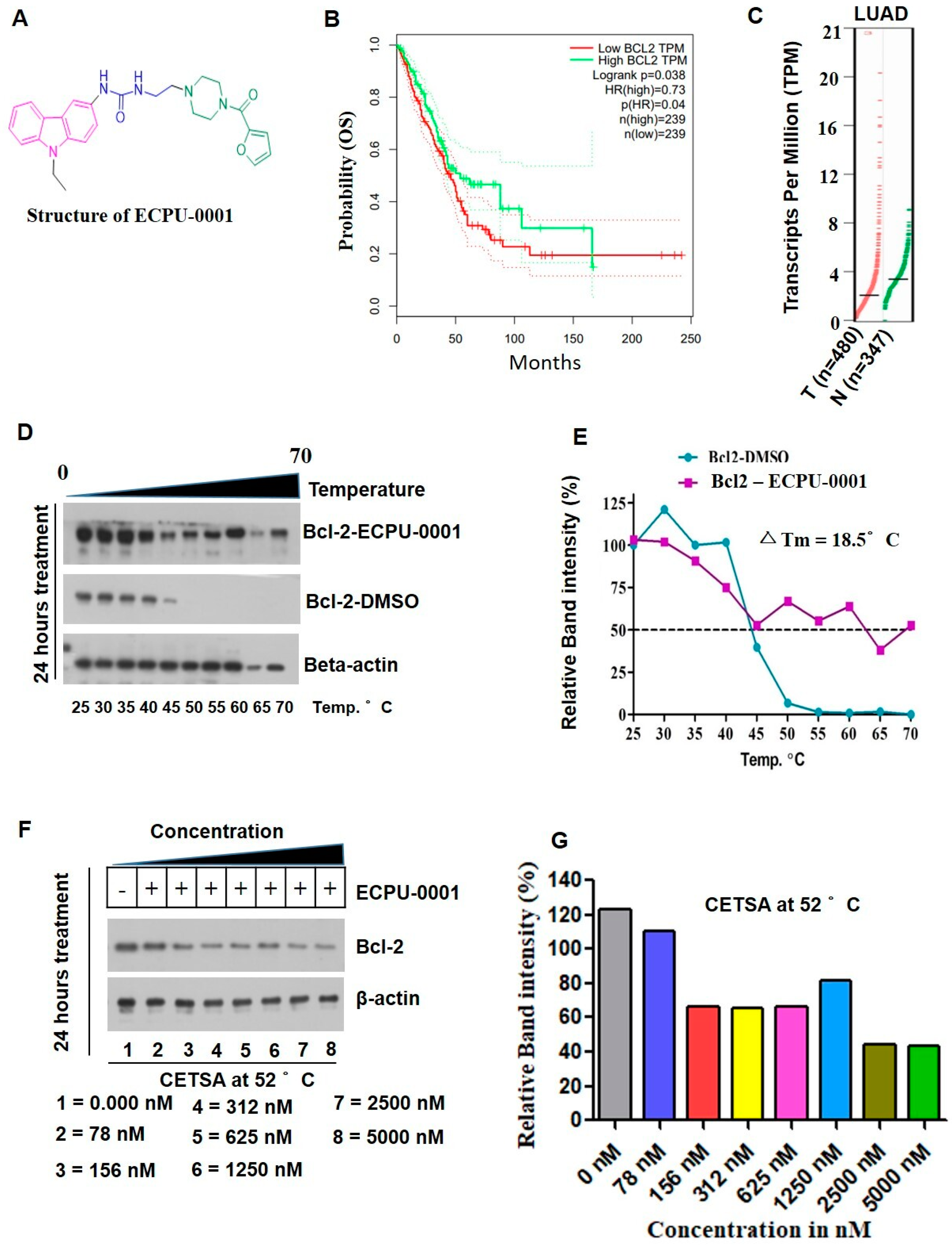
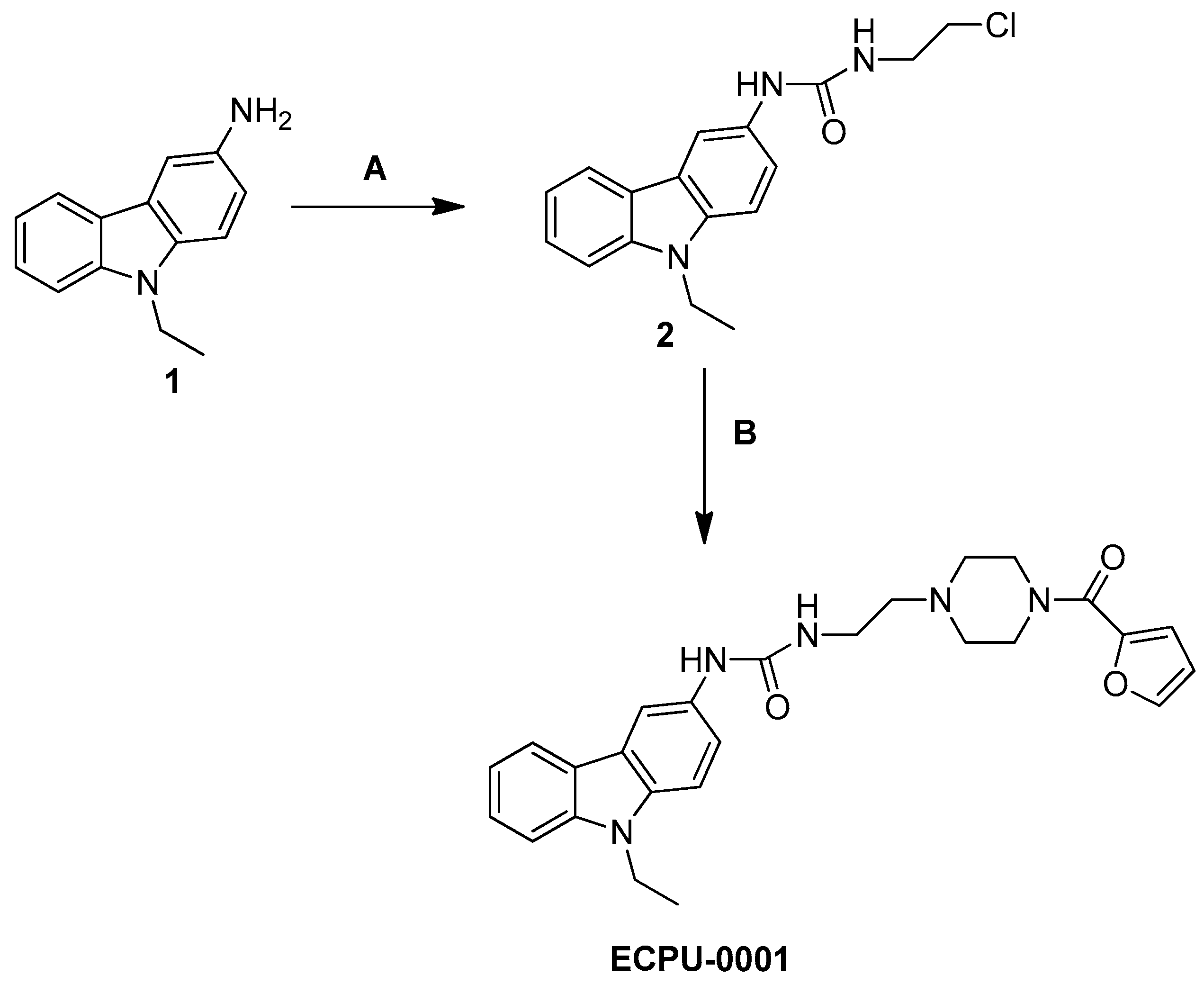
2.2. Synthesis of Novel Carbazole-Piperazine Hybrid Molecule (ECPU-0001)
2.3. ECPU-0001 Appeared as a Strong BCL-2 Targeting Agent In Vitro Study
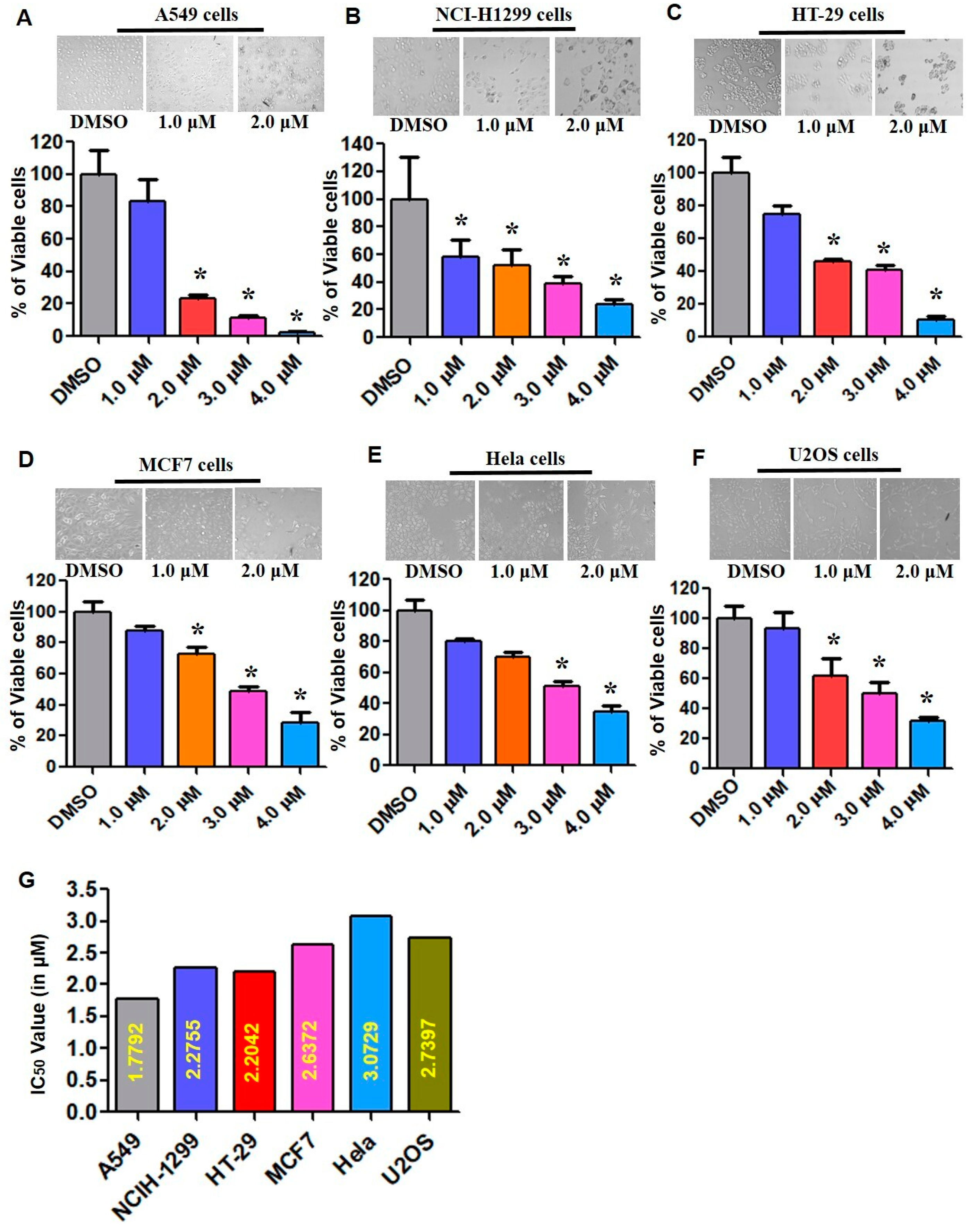
2.4. Cytotoxic Potential of ECPU-0001 in Cancer Cell Lines
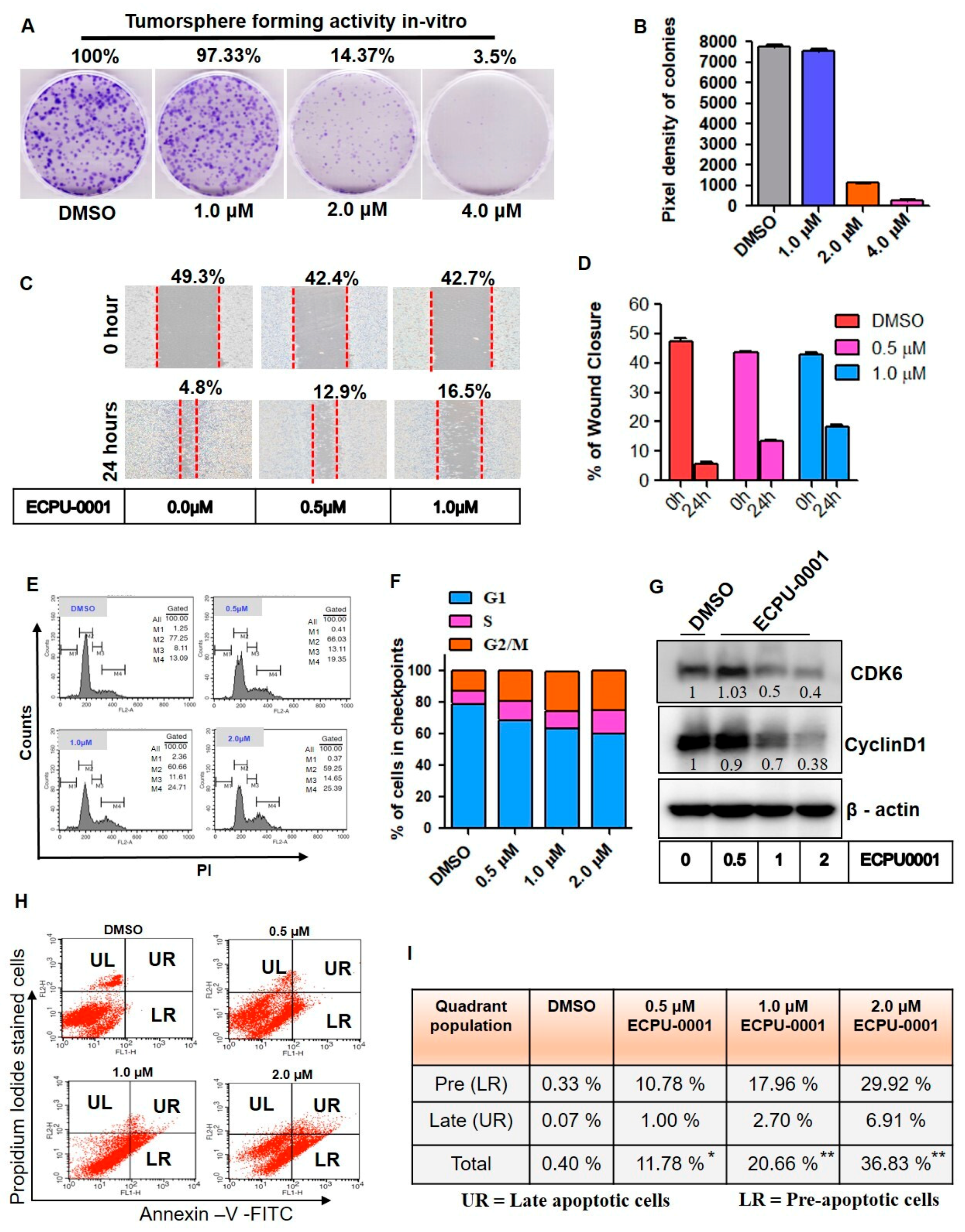
2.5. ECPU-0001 Impedes Cancer Growth and Migration In Vitro
2.6. ECPU-0001 Augments CyclinD1/CDK6 Mediated Cell Cycle Arrest and Apoptosis
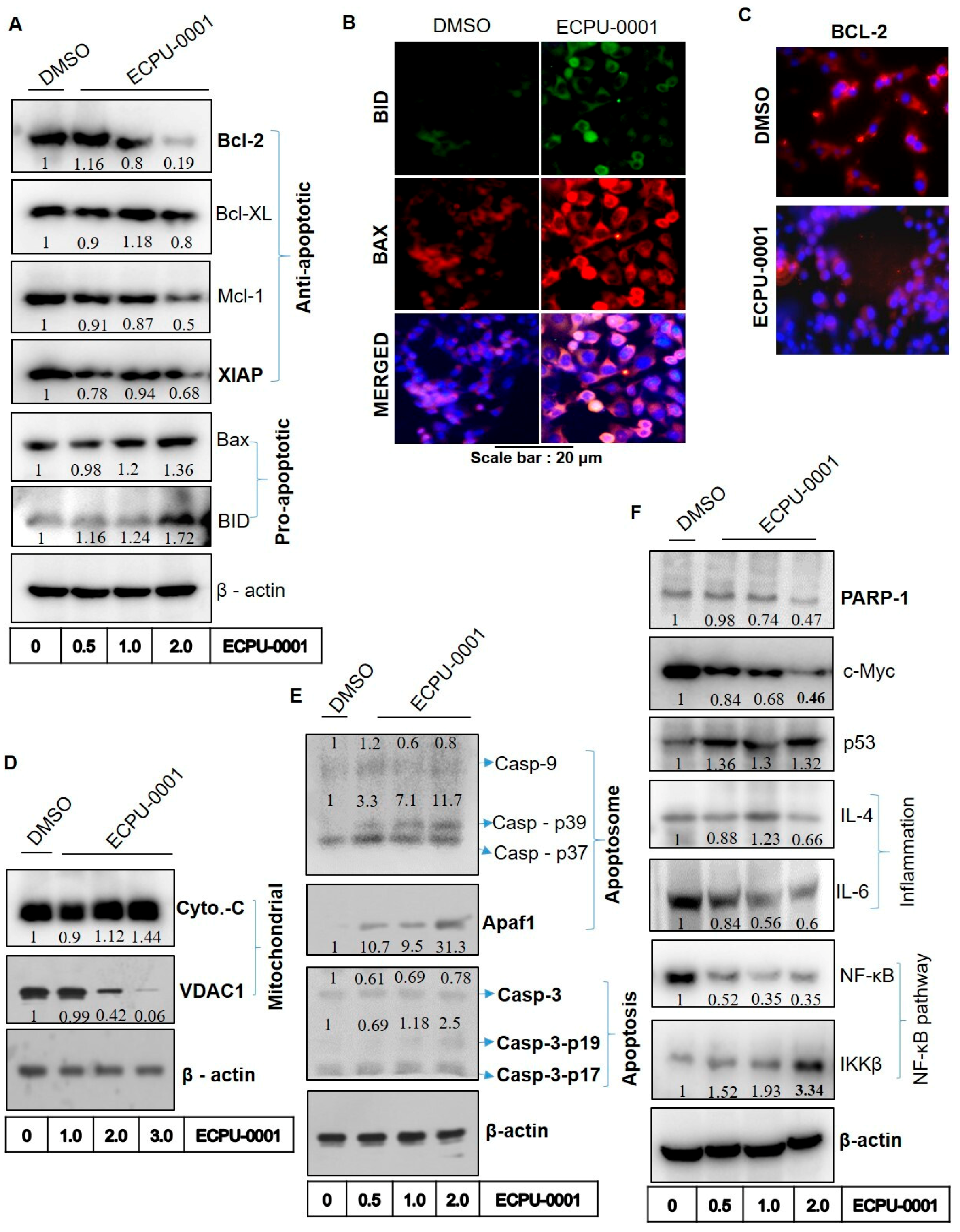
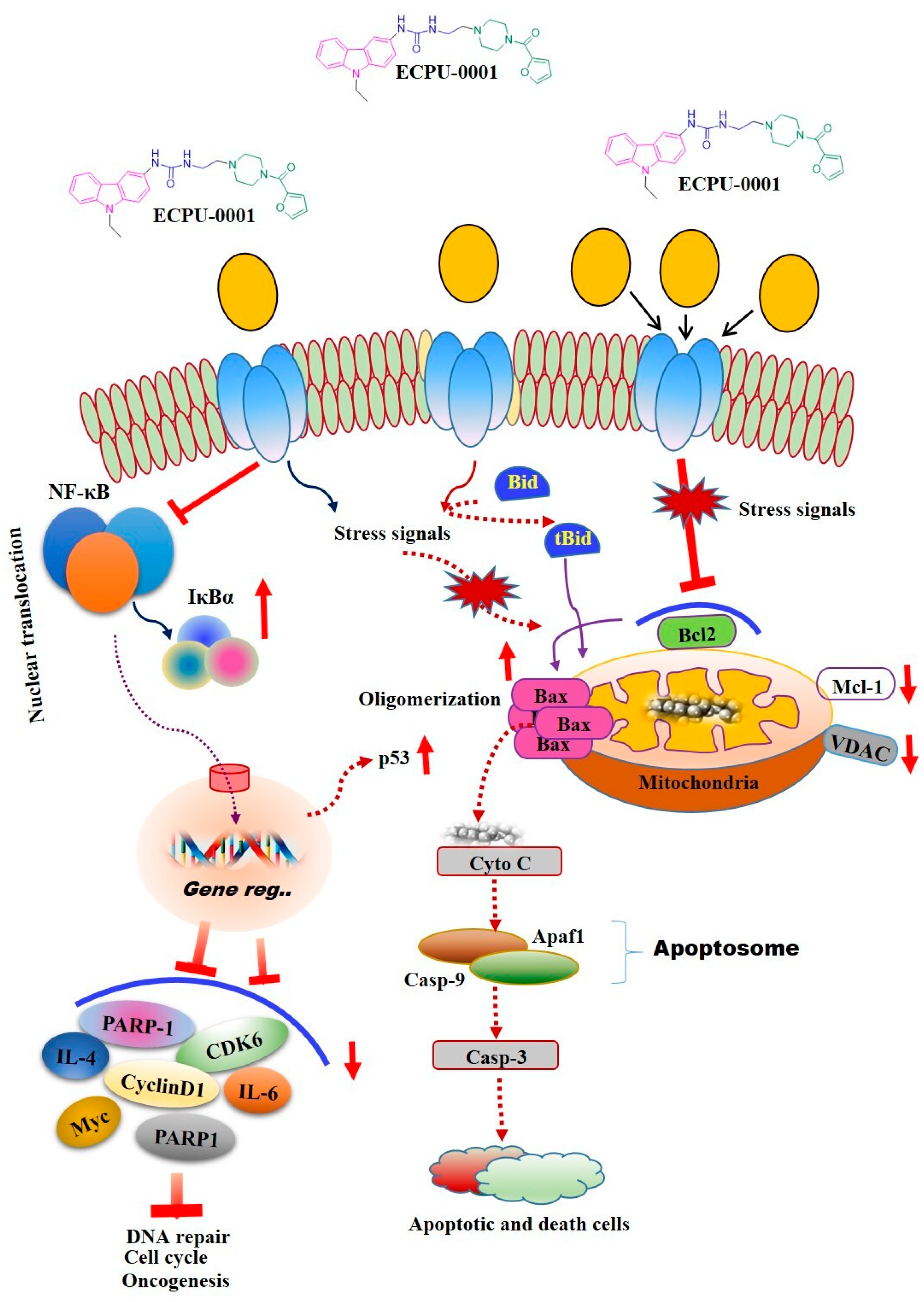
2.7. ECPU-0001 Induces Apoptosis through the Mitochondria Mediated Intrinsic Pathway by Targeting BCL-2
2.8. ECPU-0001 Inhibits A549 Cells Growth via Regulation of p53, PARP-1 and c-MYC
2.9. Effects of ECPU-0001 in Regulation of Oncogenic NF-κB, IL-4 and IL-6
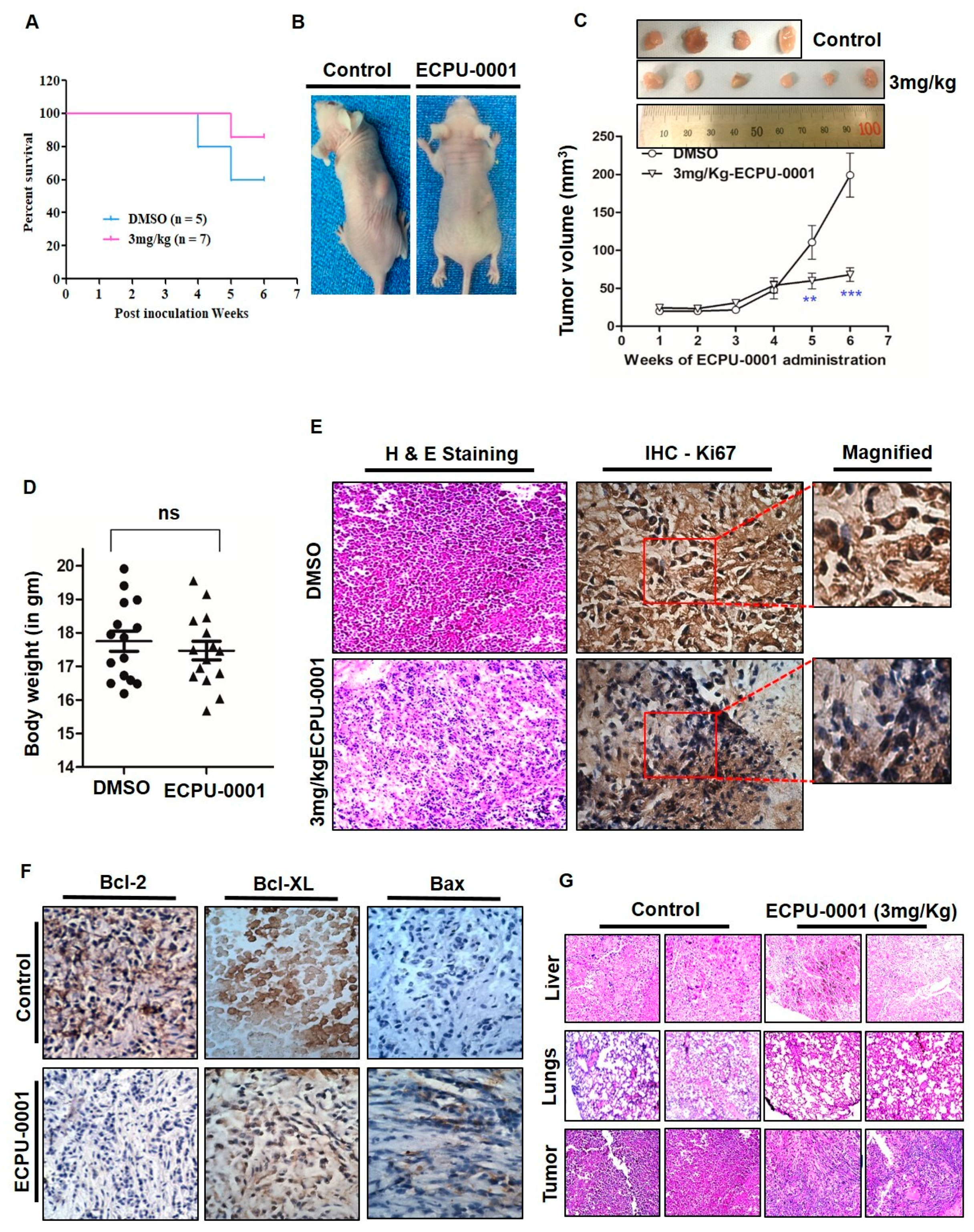
2.10. ECPU-0001 Inhibits Tumor Growth and Reduces Tumor Burden in a Xenograft Nude Mouse Model
2.11. Histopathological Investigation of Xenograft Tumor
2.12. Fabulous Tumoricidal Activity via Induction of Apoptosis In Vivo
2.13. Toxicity Evaluation
3. Discussion
4. Materials and Methods
4.1. Docking Study
4.2. Molecular Dynamics (MD) Simulation Analysis
4.3. Chemistry: Synthesis and Characterization of ECPU-0001
4.3.1. Procedure for the Synthesis of 1-(2-Chloro-ethyl)-3-(9-ethyl-9H-carbazol-3-yl)-urea(2)
4.3.2. 1-(2-Chloro-ethyl)-3-(9-ethyl-9H-carbazol-3-yl)-urea (2)
4.3.3. Procedure for the Synthesis of 1-(9-Ethyl-9H-carbazol-3-yl)-3-{2-[4-(furan-2-carbonyl)-piperazin-1-yl]-ethyl}-urea (ECPU-0001)
4.3.4. 1-(9-Ethyl-9H-carbazol-3-yl)-3-{2-[4-(furan-2-carbonyl)-piperazin-1-yl]-ethyl}-urea (2)
4.4. Cell Lines and Culture Conditions
4.5. Cellular Thermal Shift Assay (CETSA) and BCL-2-Targeting Assay
4.6. Cytotoxicity and Proliferation Assay
4.7. Early and Late Apoptosis Analysis
4.8. Cell Cycle Checkpoints Analysis by FACS
4.9. Immunocytochemistry
4.10. Protein Extraction and Immunoblot Analysis
4.11. Xenograft Development and ECPU-0001 Efficacy in Tumor-Bearing Nude Mice
4.12. Migration and Wounding Healing Assay
4.13. Immunohistochemical (IHC) Study
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NSCLC | Non-small cell lung cancer |
| APAF1 | Apoptotic protease activating factor 1 |
| XIAP | X-linked inhibitor of apoptosis protein |
| CDK6 | Cyclin-dependent kinase 6 |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| DMSO | Dimethyl sulfoxide |
| CETSA | Cellular thermal shift assay |
| VDAC1 | Voltage-dependent anion-selective channel 1 |
| IKKβ | inhibitor of nuclear factor kappa-B kinase subunit beta |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Clinical Lung Cancer Genome Project (CLCGP), Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci. Transl. Med. 2013, 5, 209ra153. [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Karachaliou, N. Lung cancer: Maintenance therapy and precision medicine in NSCLC. Nat. Rev. Clin. Oncol. 2013, 10, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Sasho, Z.P. Molecular biology of the lung cancer. Radiol. Oncol. 2005, 39, 197–210. [Google Scholar]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Katerina, P.; Roy, S.H. Lung Cancer in the Era of Precision Medicine. Clin. Cancer Res. 2015, 21, 2214–2220. [Google Scholar]
- Lockshin, R.A.; Zakeri, Z. Programmed cell death and apoptosis: Origins of the theory. Nat. Rev. Mol. Cell Biol. 2001, 2, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y. The therapeutic promise of apoptosis. Science 2019, 363, 1050–1051. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. Clearance of apoptotic cells: Implications in health and disease. J. Cell Biol. 2010, 189, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Shawgo, M.E.; Shelton, S.N.; Robertson, J.D. Caspase-mediated BAK activation and Cytochrome-C release during intrinsic apoptotic cell death in Jurkat cells. J. Biol. Chem. 2008, 283, 35532–35538. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Roberts, A.W.; Colman, P.M.; Adams, J.M. Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends Cancer 2016, 2, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Zinn, R.L.; Gardner, E.E.; Dobromilskaya, I.; Murphy, S.; Marchionni, L.; Hann, C.L.; Rudin, C.M. Combination treatment with ABT-737 and chloroquine in preclinical models of small cell lung cancer. Mol. Cancer 2013, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the BH3 mimetic BCL-2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef] [PubMed]
- Bérubé, G. An overview of molecular hybrids in drug discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef]
- Fizazi, K.; Le-Maitre, A.; Hudes, G.; Berry, W.R.; Kelly, W.K.; Eymard, J.C.; Logothetis, C.J.; Pignon, J.P.; Michiels, S. Addition of estramustine to chemotherapy and survival of patients with castration-refractory prostate cancer: A meta-analysis of individual patient data. Lancet Oncol. 2007, 8, 994–1000. [Google Scholar] [CrossRef]
- Asche, C.; Demeunynck, M. Antitumor carbazoles. Anticancer Agents Med. Chem. 2007, 7, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Głuszyńska, A. Biological potential of carbazole derivatives. Eur. J. Med. Chem. 2015, 94, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E. Ellipticines as DNA-targeted chemotherapeutics. Curr. Med. Chem. 2014, 21, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Pei, C.; He, Q.; Liang, S.; Gong, X. Mahanimbine Exerts Anticancer Effects on Human Pancreatic Cancer Cells by Triggering Cell Cycle Arrest, Apoptosis, and Modulation of AKT/Mammalian Target of Rapamycin (mTOR) and Signal Transducer and Activator of Transcription 3 (STAT3) Signalling Pathways. Med. Sci. Monit. 2018, 24, 6975–6983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Larock, R.C. Synthesis of Carbazoles and Dibenzofurans via Cross-Coupling of o-Iodoanilines and o-Iodophenols with Silylaryl Triflates and Subsequent Pd-Catalyzed Cyclization. Tetrahedron 2007, 63, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Anizon, F.; Belin, L.; Moreau, P.; Sancelme, M.; Voldoire, A.; Prudhomme, M.; Ollier, M.; Sevère, D.; Riou, J.F.; Bailly, C.; et al. Syntheses and biological activities (topoisomerase inhibition and antitumor and antimicrobial properties) of rebeccamycin analogues bearing modified sugar moieties and substituted on the imide nitrogen with a methyl group. J. Med. Chem. 1997, 40, 3456–3465. [Google Scholar] [CrossRef]
- Thongthoom, T.; Promsuwan, P.; Yenjai, C. Synthesis and cytotoxic activity of the heptaphylline and 7-methoxyheptaphylline series. Eur. J. Med. Chem. 2011, 46, 3755–3761. [Google Scholar] [CrossRef]
- Zeng, S.; Liu, W.; Nie, F.; Zhao, Q.; Rong, J.; Wang, J.; Tao, L.; Qi, Q.; Lu, N.; Li, Z.; et al. LYG-202, a new flavonoid with piperazine substitution, shows antitumor effects in-vivo and in vitro. Biochem. Biophys. Res. Commun. 2009, 385, 551–556. [Google Scholar] [CrossRef]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, A.; Henry, N.L. Palbociclib for the Treatment of Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 3591–3596. [Google Scholar] [CrossRef] [PubMed]
- Pavlovsky, C.; Chan, O.; Talati, C.; Pinilla-Ibarz, J. Ponatinib in the treatment of chronic myeloid leukemia and philadelphia chromosome positive acute lymphoblastic leukemia. Future Oncol. 2019, 15, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Q.; Lv, P.C.; Yan, T.; Zhu, H.L. Urea derivatives as anticancer agents. Anticancer Agents Med. Chem. 2009, 9, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Leggans, E.K.; Duncan, K.K.; Barker, T.J.; Schleicher, K.D.; Boger, D.L. A remarkable series of vinblastine analogues displaying enhanced activity and an unprecedentedtubulin binding steric tolerance: C20’ urea derivatives. J. Med. Chem. 2013, 56, 628–639. [Google Scholar] [CrossRef][Green Version]
- Barker, T.J.; Duncan, K.K.; Otrubova, K.; Boger, D.L. Potent Vinblastine C20’ Ureas Displaying Additionally Improved Activity Against a Vinblastine-Resistant Cancer Cell Line. ACS Med. Chem. Lett. 2013, 4, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Casara, P.; Davidson, J.; Claperon, A.; Le Toumelin-Braizat, G.; Vogler, M.; Bruno, A.; Chanrion, M.; Lysiak-Auvity, G.; Le Diguarher, T.; Starck, J.B.; et al. S55746 is a novel orally active BCL-2 selective and potent inhibitor that impairs hematological tumor growth. Oncotarget 2018, 9, 20075–20088. [Google Scholar] [CrossRef]
- Martinez, M.D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–97. [Google Scholar]
- Schiewer, M.J.; Mandigo, A.C.; Gordon, N.; Huang, F.; Gaur, S.; de Leeuw, R.; Zhao, S.G.; Evans, J.; Han, S.; Parsons, T.; et al. PARP-1 regulates DNA repair factor availability. EMBO Mol. Med. 2018, 10, e8816. [Google Scholar] [CrossRef]
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. p53 Loss in Breast Cancer Leads to c-MYC Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. 2019, 26, 624–638. [Google Scholar] [CrossRef]
- Mongre, R.K.; Sodhi, S.S.; Ghosh, M.; Kim, J.H.; Kim, N.; Park, Y.H.; Kim, S.J.; Heo, Y.J.; Sharma, N.; Jeong, D.K. The novel inhibitor BRM270 downregulates tumorigenesis by suppression of NF-κB signaling cascade in MDR-induced stem like cancer-initiating cells. Int. J. Oncol. 2015, 46, 2573–2585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hait, W.N. Anticancer drug development: The grand challenges. Nat. Rev. Drug Discov. 2010, 9, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of Cytochrome-C from mitochondria: A primary site for BCL-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Bratton, S.B.; Salvesen, G.S. Regulation of the APAF-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Li, P.F.; Dietz, R.; von Harsdorf, R. p53 regulates mitochondrial membrane potential through reactive oxygen speciesand induces Cytochrome-C-independent apoptosis blocked by BCL-2. EMBO J. 1999, 18, 6027–6036. [Google Scholar] [CrossRef]
- Shostak, K.; Chariot, A. EGFR and NF-κB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef]
- Nehra, R.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Crawford, A.C.; Clarke, R. BCL-2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and resistant breast cancer cells. FASEB J. 2010, 24, 2040–2055. [Google Scholar] [CrossRef]
- Vadevoo, S.M.P.; Kim, J.E.; Gunassekaran, G.R.; Jung, H.K.; Chi, L.; Kim, D.E.; Lee, S.H.; Im, S.H.; Lee, B. IL4 Receptor-Targeted Proapoptotic Peptide Blocks Tumor Growth and Metastasis by Enhancing Antitumor Immunity. Mol. Cancer Ther. 2017, 16, 2803–2816. [Google Scholar] [CrossRef] [PubMed]
- El-Hachem, N.; Haibe-Kains, B.; Khalil, A.; Khalil, A.; Kobeissy, F.H.; Nemer, G. AutoDock and AutoDockTools for Protein-Ligand Docking: Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1(BACE1) as a Case Study. Methods Mol. Biol. 2017, 1598, 391–403. [Google Scholar] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.C.J.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Prakash, A.; Idrees, D.; Haque, M.A.; Islam, A.; Ahmad, F.; Hassan, M.I. GdmCl-induced unfolding studies of human carbonic anhydrase IX: A combined spectroscopic and MD simulation approach. J. Biomol. Struct. Dyn. 2017, 35, 1295–1306. [Google Scholar] [CrossRef]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbäck, T.; Nordlund, P.; Martinez-Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Le, D.T.; Jung, S.; Quynh, N.T.N.; Sandag, Z.; Lee, B.S.; Kim, S.; Lee, H.; Lee, H.; Lee, M.S. Inhibitory role of AMP-activated protein kinase in necroptosis of HCT116 colon cancer cells with p53 null mutation under nutrient starvation. Int. J. Oncol. 2019, 54, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Cormier, N.; Yeo, A.; Fiorentino, E.; Paxson, J. Optimization of the Wound Scratch Assay to Detect Changes in Murine Mesenchymal Stromal Cell Migration After Damage by Soluble Cigarette Smoke Extract. J. Vis. Exp. 2015, 106, 53414. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongre, R.K.; Mishra, C.B.; Prakash, A.; Jung, S.; Lee, B.S.; Kumari, S.; Hong, J.T.; Lee, M.-S. Novel Carbazole-Piperazine Hybrid Small Molecule Induces Apoptosis by Targeting BCL-2 and Inhibits Tumor Progression in Lung Adenocarcinoma In Vitro and Xenograft Mice Model. Cancers 2019, 11, 1245. https://doi.org/10.3390/cancers11091245
Mongre RK, Mishra CB, Prakash A, Jung S, Lee BS, Kumari S, Hong JT, Lee M-S. Novel Carbazole-Piperazine Hybrid Small Molecule Induces Apoptosis by Targeting BCL-2 and Inhibits Tumor Progression in Lung Adenocarcinoma In Vitro and Xenograft Mice Model. Cancers. 2019; 11(9):1245. https://doi.org/10.3390/cancers11091245
Chicago/Turabian StyleMongre, Raj Kumar, Chandra Bhushan Mishra, Amresh Prakash, Samil Jung, Beom Suk Lee, Shikha Kumari, Jin Tae Hong, and Myeong-Sok Lee. 2019. "Novel Carbazole-Piperazine Hybrid Small Molecule Induces Apoptosis by Targeting BCL-2 and Inhibits Tumor Progression in Lung Adenocarcinoma In Vitro and Xenograft Mice Model" Cancers 11, no. 9: 1245. https://doi.org/10.3390/cancers11091245
APA StyleMongre, R. K., Mishra, C. B., Prakash, A., Jung, S., Lee, B. S., Kumari, S., Hong, J. T., & Lee, M.-S. (2019). Novel Carbazole-Piperazine Hybrid Small Molecule Induces Apoptosis by Targeting BCL-2 and Inhibits Tumor Progression in Lung Adenocarcinoma In Vitro and Xenograft Mice Model. Cancers, 11(9), 1245. https://doi.org/10.3390/cancers11091245