Abstract
Identification of targetable molecular changes is essential for selecting appropriate treatment in patients with advanced lung adenocarcinoma. Methods: In this study, a Sanger sequencing plus Fluorescence In Situ Hybridization (FISH) sequential approach was compared with a Next-Generation Sequencing (NGS)-based approach for the detection of actionable genomic mutations in an experimental cohort (EC) of 117 patients with advanced lung adenocarcinoma. Its applicability was assessed in small biopsies and cytology specimens previously tested for epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) mutational status, comparing the molecular changes identified and the impact on clinical outcomes. Subsequently, an NGS-based approach was applied and tested in an implementation cohort (IC) in clinical practice. Using Sanger and FISH, patients were classified as EGFR-mutated (n = 22, 18.8%), ALK-mutated (n = 9, 7.7%), and unclassifiable (UC) (n = 86, 73.5%). Retesting the EC with NGS led to the identification of at least one gene variant in 56 (47.9%) patients, totaling 68 variants among all samples. Still, in the EC, combining NGS plus FISH for ALK, patients were classified as 23 (19.7%) EGFR; 20 (17.1%) KRAS; five (4.3%) B-Raf proto-oncogene (BRAF); one (0.9%) Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2); one (0.9%) STK11; one (0.9%) TP53, and nine (7.7%) ALK mutated. Only 57 (48.7%) remained genomically UC, reducing the UC rate by 24.8%. Fourteen (12.0%) patients presented synchronous alterations. Concordance between NGS and Sanger for EGFR status was very high (κ = 0.972; 99.1%). In the IC, a combined DNA and RNA NGS panel was used in 123 patients. Genomic variants were found in 79 (64.2%). In addition, eight (6.3%) EML4-ALK, four (3.1%), KIF5B-RET, four (3.1%) CD74-ROS1, one (0.8%) TPM3-NTRK translocations and three (2.4%) exon 14 skipping MET Proto-Oncogene (MET) mutations were detected, and 36% were treatable alterations. Conclusions: This study supports the use of NGS as the first-line test for genomic profiling of patients with advanced lung adenocarcinoma.
1. Introduction
Lung cancer is the most common cause of cancer-related death worldwide [1], being frequently diagnosed in an advanced stage when curative treatment is no longer possible. Histologically, lung cancer is divided into small and non-small cell lung cancer (NSCLC). NSCLC is the most common type and includes squamous cell carcinoma, large-cell carcinoma and adenocarcinoma. Adenocarcinoma subtype accounts for more than half and is further defined according to different molecular subtypes by the identification of oncogenic drivers [2].
Over the last few decades, conventional platinum-based chemotherapy has produced only a modest increase in patient´s overall survival, reaching a plateau, with response rates around 35% and median survival time of 10–12 months [3]. Recent advances in the knowledge of NSCLC biology, especially on the discovery of oncogenic molecular changes leading to aberrant activation of intracellular signalling associated with the sustained growth of lung cancer cells, led to the development of genotype-targeted therapies, with significant improvement in patient´s outcomes. For instance, the identification of epidermal growth factor receptor (EGFR) kinase domain activating mutations, conferring sensitivity to EGFR tyrosine kinases inhibitors, changed the lung cancer treatment paradigm and contributed to increased progression-free survival (PFS) and quality of life in this subgroup of patients [4]. Targeted therapy for patients with anaplastic lymphoma kinase (ALK) translocations had a similar impact on prognosis [5]. In addition, the identification of other rare genomic alterations involving the ROS Proto-Oncogene 1 (ROS1), B-Raf proto-oncogene (BRAF), MET Proto-Oncogene (MET), Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2), and ret proto-oncogene (RET) genes led to the development of new targeted therapies, some already approved for first-line use [2,3,4,5,6]. Additionally, genomically unclassifiable (UC) patients are candidates for immunotherapy with checkpoint inhibitors [6].
Updated recommendations from the College of American Pathologists, International Association for the Study of Lung Cancer, Association for Molecular Pathology (CAP/IASCL/AMP), and European Society of Medical Oncology (ESMO) have strengthened the 2013 guidelines and suggest genomic testing for EGFR, ALK, and ROS1 for all advanced NSCLC, regardless of patients’ characteristics [7]. Besides, there is strong advise to perform broader molecular profiling for detection of rare mutations, to which targeted therapies are available or suitable for off-label treatment or clinical trials (ERBB2, MET, BRAF, KRAS, and RET) [8,9].
In the setting of lung cancer advanced disease, molecular diagnosis faces several difficulties. Foremost, both the quantity and quality of tumor tissue and nucleic acid content are critical. Tissue samples are often small histological fragments or cytological specimens obtained by bronchoscopy or transthoracic biopsy. These samples should be analyzed for histopathology, which also includes the programmed death-ligand 1 (PD-L1) test for immunotherapy selection and molecular testing [9]. The minimal conventional genomic study generally includes, sequentially or in parallel, at least, the EGFR and ALK mutational analysis. Sequential determinations require a substantial DNA amount, are time-consuming, cause sample´s exhaustion and often leads to under genotyping and treatment delay [10,11].
Next-Generation Sequencing (NGS) allows the sequencing of several genomic regions in a single test, in a single platform and even in samples with low DNA content. There are different NGS tests for whole genome, whole exome, or selected genes, depending on the application purpose. An NGS-based approach potentially provides a more sensitive and comprehensive genetic characterisation of lung cancer, which may impact the therapeutic options and patient´s prognosis.
In this study, we tested an NGS-based approach for the detection of actionable genomic mutations in a cohort of patients with advanced lung adenocarcinoma, previously tested sequentially for EGFR and ALK mutational status, and evaluated the clinical impact of the NGS-based approach. The primary endpoint was to assess the added-value of the NGS strategy over the sequential approach and the concordance regarding the EGFR status. Additionally, the results from the clinical implementation of a combined NGS DNA plus RNA panel are presented.
2. Material and Methods
2.1. Study Design
A total of 240 patients with advanced lung adenocarcinoma, diagnosed in the Pulmonology Department of Centro Hospitalar Universitário de São João (CHUSJ), EPE were enrolled in this study and divided into two groups: (i) experimental cohort (EC), corresponding to patients diagnosed between January 2015 and December 2016. Of a total of 127 patients previously examined for EGFR and ALK mutational status, 117 had tumor samples available for NGS retest. Ten patients were excluded from the study, seven owing to insufficient tumor sample, two with stage IIIA, and one included in another trial; (ii) implementation cohort (IC), corresponded to 123 patients diagnosed from September 2017 to July 2018, to whom NGS was integrated into the daily clinical practice (Figure 1).
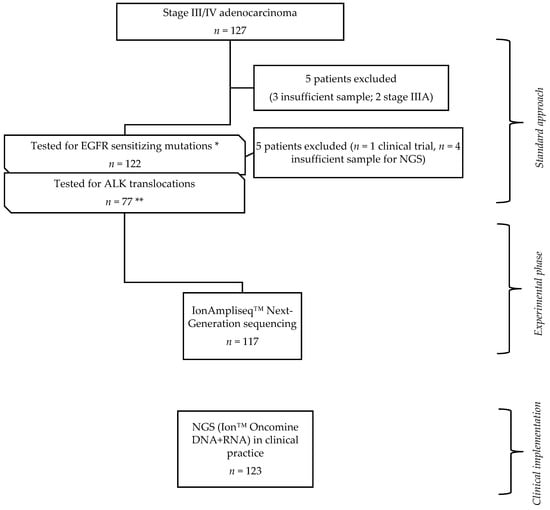
Figure 1.
Study design; 127 patients genomically profiled with sanger and anaplastic lymphoma kinase (ALK) FISH test (standard approach) were selected. Among those, in 117 the same tumor sample previously tested was submitted to Next-Generation Sequencing (NGS), comprising the experimental phase of the study (experimental cohort (EC)). Among the EC, * 22/117 were epidermal growth factor receptor (EGFR) mutated and 95 had indication for anaplastic lymphoma kinase (ALK) testing, ** 17/95 patients did not perform it due to insufficient sample. After the EC, a combined DNA + RNA panel was applied to characterize genomically patients with lung adenocarcinoma; 123 cases were included for the purpose of this study (clinical implementation phase).
The tumor staging was based on the 7th edition of the TNM staging system until December 2017 and the 8th edition from January 2018 [12,13].
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of CHUSJ (CES-108/14).
2.2. Tumor Specimens
Biopsy and cytology specimens, from the primary tumor and metastatic sites, were reviewed by pathologists. Histological specimens were fixed with formalin (formalin-fixed paraffin-embedded tissue, FFPE) and cytological specimens as smears or cellblocks. After pathological and immunohistochemical evaluation, samples were used for DNA extraction using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), following manufacturer´s instructions. DNA was quantified with NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific™, Waltham, MA, USA) or Qubit® 2.0 Fluorometer (Invitrogen, Waltham, MA, USA). All genetic analyses were done at IPATIMUP, a College of American Pathologists and ISO15189 accredited laboratory.
2.3. Library and Template Preparation for Next-Generation Sequencing
The Ion AmpliSeq Colon and Lung Cancer Research Panel v2 (Ion Torrent, Waltham, MA, USA) was used to detect changes in DNA, in the experimental phase. This multiplex PCR-based test allows the analysis of 1850 hotspots and targeted regions in 22 genes (AKT1, ALK, BRAF, CTNNB1, DDR2, EGFR, ERBB2, ERBB4, FBX7, FGFR3, FGFR1, FGFR2, KRAS, MAP2K1, MET, NOTCH1, NRAS, PTEN, PIK3CA, STK11, SMAD4 and TP53) involved in tumorigenesis. Libraries were generated using 1–10 ng of DNA from tissue FFPE blocks sections, according to the manufacturer.
In the clinical implementation phase, samples were characterised using the Oncomine Solid Tumor kits CE-IVD (Ion Torrent, Waltham, MA, USA). These assays allow the analysis of the same targets as the Ion AmpliSeq Colon and Lung Cancer Research Panel v2 plus the detection of ALK, RET, ROS1, and NTRK1 gene fusions. Libraries were generated using 1–10 ng of DNA and RNA from tissue FFPE blocks sections, according to the manufacturer. The final libraries were quantified by qPCR with the Ion Library TaqMan® Quantitation Kit (Ion Torrent, Waltham, MA, USA) and used for template preparation performed using the Ion Chef (Ion Torrent, Waltham, MA, USA).
2.4. Next-Generation Sequencing and Bioinformatic Analysis
Loaded chips were sequenced in an Ion PGM or Ion S5XL sequencer. Sequencing quality was assessed through the plug-in coverage analysis and the samples were analyzed using dedicated bioinformatic workflows within the Ion Reporter v5.6 server (Ion Torrent, Waltham, MA, USA). Samples with a number of reads <100,000 and/or the average base coverage <500× were considered inadequate for analysis. The amplicons with a coverage <250× were considered non-informative. Mutations with allele frequencies of at least 10% and adequate coverage in target regions were considered to call a mutation in a patient sample. Polymorphisms, synonymous or intronic mutations were excluded. The Catalogue of Somatic Mutations in Cancer (COSMIC) database was used to access the clinically relevant variants. Some mutations detected by NGS were validated by digital PCR.
2.5. Statistical Analysis
The sample size was limited by the availability of specimens for subsequent NGS analysis following sequential standard molecular diagnostic approach. Most of the analysis was descriptive. Categorical data were described as absolute (n) and relative frequencies. Medians, interquartile ranges (IQR), and minimum and maximum values were determined for continuous variables. The NGS results were correlated with other parameters and assessed with the chi-square test or Fisher´s exact test, when appropriate. Cohen´s Kappa test was used to assess the inter-rater agreement for categorical data. Kaplan-Meier actuarial curve analysis was used to assess survival and the log-rank test for the chi-square (X2) calculus, for each event time and each evaluated group. The significance level assumed was 0.05. All statistical analyses were performed using the Statistical Package for Social Sciences (SPSS, IBM Corp, Chicago, IL, USA) software, version 25.0.
3. Results
3.1. Clinicopathologic Characteristics and Mutational Profile
In this study, 117 and 123 patients were, respectively, included in the EC and IC groups. The clinicopathologic features are presented in Table 1. All specimens were adenocarcinoma, except one suggestive of adenosquamous carcinoma (in the EC). Most of patients had stage IV disease at the time of diagnosis. In both groups, samples were predominantly core biopsies suitable for histological analysis. Cytological samples were obtained by fine-needle-aspiration techniques, as pleural and pericardial fluid aspiration, endobronchial ultrasound-guided needle aspiration (EBUS-TBNA), and lung and peripheral lymph nodes fine-needle aspirations (Table 1).

Table 1.
Patient´s demographics and clinical characteristics.
NGS analysis succeeded in both histological and cytological samples, with no statistically significant differences between groups (p = 0.487). The frequency of alterations found did not differ in both histological and cytological samples between both cohorts. In the EC, 7/21 mutations were from cytological samples and 49/96 in histological samples (p = 0.141). In the IC, hotspot alterations were found in 12/18 cytological and 66/105 histological samples (p = 0.538).
In the EC, the EGFR mutation test was performed in all patients, with a positive rate of 18.8% (22/117). ALK translocations were identified in 7.7% of cases (9/117) (Table 1). The remaining cases were designated as UC (n = 86, 73.5%). Of the 95 EGFR negative cases with ALK FISH testing indication, 17.9% (17/95) had insufficient sample for ALK test. EGFR-mutated patients were distributed as follows: 16 (72.7%) exon19 deletions (19 DEL), five (22.7%) L858R, and one (4.5%) exon20 insertion (20 Ins) (Table S1).
3.2. Next-Generation Sequencing Results
According to the results obtained with the Ion AmpliSeqTM Colon and Lung Cancer Research Panel v2 for DNA analysis, 56 (47.9%) patients harbored at least one gene variant and in 61 (52.1%) no genomic alteration was identifiable, remaining as UC. Analyzing by patient and comparing with the standard approach (Figure 2A), the most frequent alterations were KRAS (n = 23, 19.7%), EGFR (n = 23, 19.7%), BRAF (n = 5, 4.3%), TP53 (n = 3, 2.6%), ERBB2 (n = 1, 0.9%), and STK11 (n = 1, 0.9%). Analyzing by sample, a total of 68 genomic variants were identified (Figure 2B; Table S2).
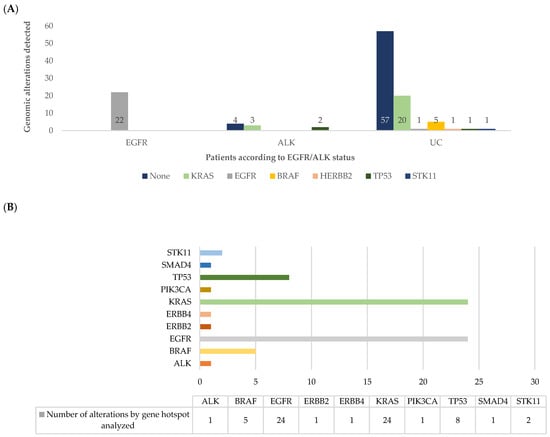
Figure 2.
(A) Number of patients with genomic alterations assessed by NGS findings, in each patient´s group assessed by the standard classification (EGFR, ALK, and unclassifiable (UC)); (B) number of hotspot alterations in each gene analyzed by the Ion AmpliSeq™ assay.
Of the 86 (73.5%) patients classified as UC by the standard approach, 29 (33.7%) had at least one gene alteration. Beyond EGFR mutations, variants were observed in other targetable genes, as described above. Two additional EGFR mutations were found, one not previously identified and one as co-alteration.
Combining NGS for DNA analysis data plus ALK detection by FISH, 60 (51.3%) patients had at least one alteration identified, and 57 (48.7%) remained without identified mutations, reducing the UC rate by 24.8% (Figure 3A,B).
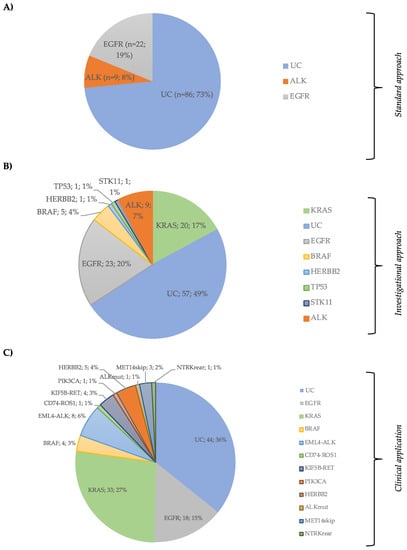
Figure 3.
(A) Patient’s distribution by EGFR and ALK status (Standard Approach); (B) patient’s classification by Ion Ampliseq Lung and colon panel plus ALK FISH status (Investigational Approach); (C) distribution of hotspot DNA and RNA subgroups in the clinical cohort (Clinical application of the combined DNA + RNA Panel).
3.3. Concurrent Genomic Alterations
Concurrent changes were found in 14 patients (12.0%) (Table 2), with emphasis on co-alterations occurring in EGFR (4/23) and ALK (5/9) mutated patients. Among the EGFR mutated patients, one exhibited an ALK point mutation c.3512T>A in a neglectable percentage; one patient, with a combination of Del19 with allelic frequency of 66% plus p.T790M with allelic frequency of 0.6%, had a PFS with a 1st generation TKI of 9.2 months and progressed with the p.T790M point mutation; the other two patients had a KRAS mutation and a PIK3CA mutation, respectively. In the ALK-positive patients’ subgroup, a molecular co-alteration was detected in five cases: three with KRAS and two with TP53 mutations. Two of the ALK plus KRAS mutated patients had a very dismal evolution with overall survival (OS) less than 3 months (Table 2).
Table 2.
Cases with concomitant genomic alterations.
3.4. Concordance Between Sanger and NGS for the EGFR Status
A set of 117 patients was analyzed by both Sanger sequencing and NGS for EGFR mutations detection, allowing the concordance determination between tests. Overall, the percentage of concordant cases was 116/117 (99.1%) (Table 3), and there was an almost perfect agreement between the two tests (κ = 0.972; p < 0.001).
Table 3.
Comparison between NGS and Sanger for EGFR status.
Assuming that all EGFR positive cases are true positives, in this cohort, 23 patients were EGFR mutated. The sensitivity for Sanger was 95.6% and for NGS 100%. One extra EGFR mutated patient was found among the unclassified population by Sanger.
3.5. Impact of the Identification of Targetable Alterations in Patient’s Overall Survival
The EC presented a median OS of 13.0 months (95% CI 8.1–17.9, sd = 2.5): 24 months (95% CI 10.2–37.8, sd = 7.0) for the EGFR subgroup, 11 months (95% CI 5.2–16.8, sd = 2.98) for ALK, and 11 months for UC patients (95% CI 5.98–16.0, sd = 2.6). Specific survival patterns were observed in subtypes of oncogenic alterations detected by NGS, with EGFR patients having the best median OS and KRAS the worse (Figure 4A). Among patients with a druggable oncogenic alteration, 30 were treated with targeted therapy, 21/23 EGFR, 2/5 BRAF, 7/9 (ALK) were treated with TKIs. Regardless of the treatment line, OS was significantly higher for this subgroup of patients (median OS 24 vs. 9 months, p = 0.028) (Figure 4B). As groups were small, no statistical comparisons were feasible.
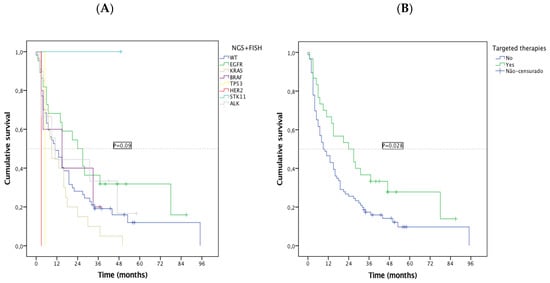
Figure 4.
Kaplan-Meier overall survival (OS) by (A) NGS subgroups; (B) targeted treatment vs. non-targeted treatment.
3.6. NGS Results from the Implementation Cohort with a Combined DNA and RNA Panel
Combined DNA and RNA sequencing succeeded in 117 (95.1%). DNA degradation occurred in four (3.2%) cases and in these cases, EGFR was analyzed by Sanger sequencing. RNA extraction failed in two cases (1.6%), with no statistically significant differences in the sequence rate according to the sample type (16/18 for cytology and 102/105 for histology, p = 0.1).
DNA and RNA hotspot alterations were detected in 79 (64.2%) patients. KRAS mutations occurred in 33 (26.8%) patients, EGFR in 18 (14.6%), BRAF in four (3.2%), ERBB2 in five (4.1%), and PIK3CA in one (0.8%). Exon 14 skipping MET mutation was detected in three (2.4%) patients. Considering gene translocations, eight (6.5%) patients had an ELM4-ALK, four (3.1%) an KIF5B-RET, one (0.8%) an CD74-ROS1, and one (0.8%) an TPM3-NTRK (Figure 3C and Table S3). Only 44 (35.8%) patients remained as UC. Forty-four (35.8%) patients had treatable alterations. Additionally, co-alterations were found in 11 (8.9%) cases (Table S4).
4. Discussion
Lung cancer treatment is rapidly evolving from histology to precision-based therapy, relying on predictive biomarkers determined by molecular tests. Sequential analysis of each gene with single-gene assays is unable to search efficiently for all actionable mutations, due to tumor sample extinction. For completing the minimum genomic characterisation, patients would have to undergo additional diagnostic procedures, with increased risk and costs. Testing each biomarker, one at a time, may result in longer and potentially unacceptable turnaround time. Besides, and considering the number of genes that must be tested, it will probably not be cost-effective.
Lung cancer diagnosis is established predominantly in small biopsies or cytological samples, which must be enough for morphology, immunohistochemistry (IHC), and molecular evaluation, essential to determine if an actionable oncogenic alteration is present. In real-world clinical practice, many obstacles arise to meet this need. The barriers consist of cost, number of separate assessments required, quality of tumor samples and local availability of the tests [14,15].
In the last years, Sanger sequencing has been considered the gold standard for gene mutation testing, particularly, for EGFR. However, this technique is not very sensitive, requires samples with at least 20% of tumor cells and its application to small lung biopsies or cytology specimens is not always possible. In addition, to assess ALK rearrangements, the gold standard is IHC or FISH, both tests needing many histological sections from an FFPE or a cellblock.
High-throughput NGS, also referred to as massively parallel sequencing, allows multiplex PCR with simultaneous amplification of a pre-specified panel of genes in a single reaction. Depending on the panel, it offers the possibility to analyze DNA, RNA, transcription regions, methylation patterns and, more recently, tumor mutation burden. Targeted gene panels represent an alternative method for capturing specific genomic regions for subsequent sequencing and is the best option for molecular characterization of lung cancer, allowing multiple genes to be analyzed at the same time with enough depth of coverage to detect minor allele frequencies. Most NGS assays require as little as 10 ng of DNA, or even less, while non-NGS tests, like Sanger, require more DNA. NGS was previously validated in several studies and its superiority is recognised in terms of sensitivity, speed, and costs [16,17,18,19].
In the present study, we report our experience with a targeted NGS-based strategy, using the Thermofisher Ion Torrent NGS technology, with the Ion AmpliSeq Colon and Lung Cancer Research Panel v2, for the detection of actionable genomic alterations in a cohort of patients with lung adenocarcinoma, and the steps taken until its prospective integration into our clinical practice. This panel was previously validated in a study, in which the same sample was tested in seven different laboratories of the OncoNetwork Consortium [20]. In our study, tumor specimens were previously analyzed for the determination of EGFR and ALK mutational status, by Sanger sequencing and FISH, respectively, according to recommendations at that time [7]. After morphological and molecular standard subtyping, samples were retested with the targeted-gene NGS panel. NGS was done in samples with an average of 5.1 ng DNA, suggesting that successful sequencing may occur in samples with small DNA amounts, overcoming the difficulty of mutation analysis in the context of lung cancer diagnosis [15].
In our cohort, only 77 (65.8%) patients had both EGFR and ALK genes analyzed, reflecting the difficulty in completing the minimal recommended genomic analysis, based on single sequential tests, as previously mentioned. The specimens were diverse and the Ion Ampliseq sequencing proved to be effective even in critical samples, such as those obtained by FNA and biological fluids. Scarpa et al. [21] demonstrated, for the first time, the successful application of the Ion AmpliSeq Colon and Lung Cancer in cytological samples of lung adenocarcinoma, and these results were replicated in other studies [14,22,23,24].
NGS revealed excellent accuracy for EGFR detection and a very high concordance rate (99.1%) with Sanger sequencing. There was only one discordant case, an EGFR deletion (EGFR c.2236_2250del15) detected with NGS but missed by Sanger. Indeed, previously published studies have also highlighted the NGS superiority over the traditional methods [25,26,27]. Although not an epidemiological study, mutation patterns and frequencies agree with previous data published on our population [28].
Our data demonstrated high sensitivity to detect druggable alterations, significantly reducing the rate of genomically unclassifiable patients to 48.7%, with the combination of the Ion Ampliseq panel and ALK FISH test. Additionally, we identified molecular changes in 29/86 (33.7%) of the UC patients classified by the standard approach, as reported in similar studies [29,30]. Beyond KRAS mutations, one more patient with an EGFR activating mutation and five with BRAF-p.V600E could have been treated with targeted agents or included in clinical trials.
NGS also provided some insights into the biology and clinical behavior of the disease. According to the survival analysis, subsets of patients could be defined: EGFR-mutated patients presented higher median OS and KRAS the lowest ones, which is similar to the literature data [31,32]. Unexpectedly, ALK patients had a low OS, perhaps due to the unavailability of TKIs as first-line treatment at the beginning of the study and to the coexistence of KRAS mutation in three of the nine ALK patients. This occurrence is rare, but it has already been reported as being associated with primary resistance to TKIs [32,33]. The expected co-occurrence of TP53 mutations with KRAS, EGFR, BRAF, PIK3CA, and STK11 were also identified. Another interesting finding was the detection, before treatment, of a low allelic frequency p.T790M point mutation alongside the driver Del19, which, in this case, prompted progression through this on-target resistance mechanism. The identification of simultaneous alterations can explain the unexpected clinical outcomes and resistance patterns, suggesting the existence of subclonal populations or hypothetically a different population of tumor cells. Considering the small sample size of this study, definitive conclusions on the clinical impact of a concurrent genomic alteration cannot be driven and deserve further investigation.
After the evaluation phase of this DNA panel, since it has limitations regarding chromosomal rearrangements detection, a combined DNA and RNA panel, Ion TorrentTM Oncomine™ Focus Assay [34], was integrated into the lung cancer molecular diagnosis. This panel allowed even a higher reduction of unclassified patients to 36%. In this clinical cohort, beyond the target point mutations and deletions found, exon 14 skipping MET mutation and gene translocations, ELM4-ALK, KIF5B-RET, CD74-ROS1, and even a TPM3-NTRK were detected. Overall, in the clinical cohort, and considering drug’s availability, about 36% of patients were candidates for specific therapy. These data demonstrate that this combined targeted NGS sequencing methodology allows the detection of common and some clinically relevant rare mutations. This strategy can identify more patients who are likely to benefit from specific therapies, improving clinical outcomes, minimising the toxic effects, and promoting drug development.
Our data are equivalent to those obtained in larger studies. In the Lung Cancer Consortium, an actionable oncogenic alteration was found in 64% of patients [30]. In a study made by Foundation Medicine, 6.832 clinical NSCLC samples were tested with a comprehensive hybridization capture panel with more than 300 genes, able to detect also translocations, and 71% of patients harbored at least one genomic alteration [35].
This study was practice changing. The routine diagnostic workup is, at present, a reflex test including IHC, PDL1, and a combined DNA and RNA next-generation sequencing on FFPE tissue or cytological samples, for all patients with NSCLC with genomic testing indication. This analysis was mainly focused on the clinical usefulness of NGS, in search of suitable patients for targeted approved drugs, off-label treatments and clinical trials, but the spectrum of applications goes further beyond that. This kind of combined panel, profiling DNA and RNA, has also the potential to detect alterations that may soon have available drugs and the ability to identify and further characterize resistance mechanisms and aberrant clinical patterns. Future challenges will be the interpretation of co-alterations, low-frequency variants, and those of uncertain biological meaning.
5. Conclusions
This study contributed to the clinical validation of NGS for screening actionable mutations in patients with lung adenocarcinoma, demonstrating good accuracy in biological specimens with small DNA content, like those obtained for lung cancer diagnosis. In addition, it provided a more comprehensive genomic characterisation of the disease, contributing to a better definition of tumor biology.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6694/11/9/1229/s1, Table S1: EGFR mutations assessed by Sanger, Table S2: NGS mutation annotation and allelic frequency of the experimental cohort, Table S3: NGS mutation annotation of the clinical implementation cohort, Table S4: NGS co-alterations in the clinical implementation cohort, Methods S1: Sanger sequencing, Methods S2: Fluorescence in situ hybridization (FISH).
Author Contributions
Conceptualization, M.G.O.F., J.C.M., V.H. and J.L.C.; methodology, M.G.O.F., N.M., J.C.M. and J.L.C.; software, M.G.O.F., N.M., J.P.R., A.J., M.J.P., L.C., J.L.C.; formal analysis, M.G.O.F. and N.M.; investigation, M.G.O.F., M.J., C.S.M., S.G., C.S., J.P.; data curation, M.G.O.F., M.J. and N.M.; writing—original draft preparation, M.G.O.F.; writing—review and editing, M.G.O.F., N.M. and J.L.C.; supervision, J.A.M., J.C.M, V.H. and J.L.C. All authors read and approved the final version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We acknowledge the work of the members of our department, especially doctors from the thoracic oncology unit, oncology and pulmonology nurses and, patients and their relatives. N. Martins would like to thank the Portuguese Foundation for Science and Technology (FCT-Portugal) for the Strategic project ref. UID/BIM/04293/2013 and "NORTE2020—Northern Regional Operational Program" (NORTE-01-0145-FEDER-000012).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Shames, D.S.; Wistuba, I.I. The Evolving Genomic Classification of Lung Cancer. J. Pathol. 2014, 232, 121–133. [Google Scholar] [CrossRef]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison of Four Chemotherapy Regimens for Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Thorac. Oncol. 2013, 8, 823–859. [Google Scholar]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar]
- Riely, G.L. What, When, and How of Biomarker Testing in Non-Small Cell Lung Cancer. J. Natl. Compr. Canc. Netw. 2017, 15, 686–688. [Google Scholar] [CrossRef]
- Lim, C.; Tsao, M.S.; Le, L.W.; Shepherd, F.A.; Feld, R.; Burkes, R.L.; Liu, G.; Kamel-Reid, S.; Hwang, D.; Tanguay, J.; et al. Biomarker testing and time to treatment decision in patients with advanced nonsmall-cell lung cancer. Ann. Oncol. 2015, 26, 1415–1421. [Google Scholar] [CrossRef]
- Gutierrez, M.E.; Choi, K.; Laan, R.B.; Licitra, E.J.; Skrzypczak, S.M.; Pe Benito, R.; Wu, T.; Arunajadai, S.; Kaur, S.; Harper, H.; et al. Genomic Profiling of Advanced Non-Small Cell Lung Cancer in Community Settings: Gaps and Opportunities. Clin. Lung Cancer 2017, 18, 651–659. [Google Scholar] [CrossRef]
- Groome, P.A.; Bolejack, V.; Crowley, J.J.; Kennedy, C.; Krasnik, M.; Sobin, L.H.; Goldstraw, P. The IASLC Lung Cancer Staging Project: Validation of the proposals for revision of the T, N, and M descriptors and consequent stage groupings in the forthcoming (seventh) edition of the TNM classification of malignant tumours. J. Thorac. Oncol. 2007, 2, 694–705. [Google Scholar] [CrossRef]
- Detterbeck, F.C.; Boffa, D.J.; Kim, A.W.; Tanoue, L.T. The Eighth Edition Lung Cancer Stage Classification. Chest 2017, 151, 193–203. [Google Scholar] [CrossRef]
- Treece, A.L.; Montgomery, N.D.; Patel, N.M.; Civalier, C.J.; Dodd, L.G.; Gulley, M.L.; Booker, J.K.; Weck, K.E. FNA smears as a potential source of DNA for targeted next-generation sequencing of lung adenocarcinomas. Cancer Cytopathol. 2016, 124, 406–414. [Google Scholar] [CrossRef]
- Roy-Chowdhuri, S.; Goswami, R.S.; Chen, H.; Patel, K.P.; Routbort, M.J.; Singh, R.R.; Broaddus, R.R.; Barkoh, B.A.; Manekia, J.; Yao, H.; et al. Factors affecting the success of next-generation sequencing in cytology specimens. Cancer Cytopathol. 2015, 123, 659–668. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. JMD 2015, 17, 251–264. [Google Scholar]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. 2015, 21, 3631–3639. [Google Scholar] [CrossRef]
- D’Haene, N.; Le Mercier, M.; De Nève, N.; Blanchard, O.; Delaunoy, M.; El Housni, H.; Dessars, B.; Heimann, P.; Remmelink, M.; Demetter, P.; et al. Clinical Validation of Targeted Next Generation Sequencing for Colon and Lung Cancers. PLoS ONE 2015, 10, e0138245. [Google Scholar] [CrossRef]
- Tops, B.B.; Normanno, N.; Kurth, H.; Amato, E.; Mafficini, A.; Rieber, N.; Le Corre, D.; Rachiglio, A.M.; Reiman, A.; Sheils, O.; et al. Development of a semi-conductor sequencing-based panel for genotyping of colon and lung cancer by the Onconetwork consortium. BMC Cancer 2015, 15, 26. [Google Scholar] [CrossRef]
- Scarpa, A.; Sikora, K.; Fassan, M.; Rachiglio, A.M.; Cappellesso, R.; Antonello, D.; Amato, E.; Mafficini, A.; Lambiase, M.; Esposito, C.; et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS ONE 2013, 8, e80478. [Google Scholar] [CrossRef]
- Qiu, T.; Guo, H.; Zhao, H.; Wang, L.; Zhang, Z. Next-generation sequencing for molecular diagnosis of lung adenocarcinoma specimens obtained by fine needle aspiration cytology. Scientific Reports 2015, 5, 11317. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Portier, B.P.; Singh, R.R.; Routbort, M.J.; Aldape, K.D.; Handal, B.A.; Rahimi, H.; Reddy, N.G.; Barkoh, B.A.; Mishra, B.M.; et al. Next-generation sequencing-based multi-gene mutation profiling of solid tumors using fine needle aspiration samples: Promises and challenges for routine clinical diagnostics. Mod. Pathol. 2014, 27, 314–327. [Google Scholar] [CrossRef]
- Karnes, H.E.; Duncavage, E.J.; Bernadt, C.T. Targeted next-generation sequencing using fine-needle aspirates from adenocarcinomas of the lung. Cancer Cytopathol. 2014, 122, 104–113. [Google Scholar] [CrossRef]
- McCourt, C.M.; McArt, D.G.; Mills, K.; Catherwood, M.A.; Maxwell, P.; Waugh, D.J.; Hamilton, P.; O’Sullivan, J.M.; Salto-Tellez, M.; et al. Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PLoS ONE 2013, 8, e69604. [Google Scholar] [CrossRef]
- Tuononen, K.; Maki-Nevala, S.; Sarhadi, V.K.; Wirtanen, A.; Ronty, M.; Salmenkivi, K.; Andrews, J.M.; Telaranta-Keerie, A.I.; Hannula, S.; Lagstrom, S.; et al. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Gene. Chromosome. Cancer 2013, 52, 503–511. [Google Scholar]
- Marchetti, A.; Del Grammastro, M.; Filice, G.; Felicioni, L.; Rossi, G.; Graziano, P.; Sartori, G.; Leone, A.; Malatesta, S.; Iacono, M.; et al. Complex Mutations & Subpopulations of Deletions at Exon 19 of EGFR in NSCLC Revealed by Next Generation Sequencing: Potential Clinical Implications. PLoS ONE 2012, 7, e42164. [Google Scholar]
- Aguiar, F.; Fernandes, G.; Queiroga, H.; Machado, J.C.; Cirnes, L.; Souto Moura, C.; Hespanhol, V. Overall Survival Analysis and Characterization of an EGFR Mutated Non-Small Cell Lung Cancer (NSCLC) Population. Arch. Bronconeumol. 2018, 54, 10–17. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, E.Y.; Kim, H.R.; Ali, S.M.; Greenbowe, J.R.; Shim, H.S.; Chang, H.; Lim, S.; Paik, S.; Cho, B.C. Genomic profiling of lung adenocarcinoma patients reveals therapeutic targets and confers clinical benefit when standard molecular testing is negative. Oncotarget. 2016, 7, 24172–24178. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Pan, W.; Yang, Y.; Zhu, H.; Zhang, Y.; Zhou, R.; Sun, X. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: A meta-analysis of 41 studies. Oncotarget 2016, 7, 8373–8388. [Google Scholar] [CrossRef]
- Schmid, S.; Gautschi, O.; Rothschild, S.; Mark, M.; Froesch, P.; Klingbiel, D.; Reichegger, H.; Jochum, W.; Diebold, J.; Früh, M. Clinical Outcome of ALK Positive Non-Small Cell Lung Cancer (NSCLC) Patients with De Novo EGFR or KRAS Co-Mutations Receiving Tyrosine Kinase Inhibitors (TKIs). J. Thorac. Oncol. 2017, 12, 681–688. [Google Scholar] [CrossRef]
- De Marchi, F.; Haley, L.; Fryer, H.; Ibrahim, J.; Beierl, K.; Zheng, G.; Gocke, C.D.; Eshleman, J.R.; Belchis, D.; Illei, P.; et al. Clinical Validation of Coexisting Activating Mutations Within EGFR, Mitogen-Activated Protein Kinase, and Phosphatidylinositol 3-Kinase Pathways in Lung Cancers. Arch. Pathol. Lab. Med. 2019, 143, 174–182. [Google Scholar] [CrossRef]
- Williams, H.L.; Walsh, K.; Diamond, A.; Oniscu, A.; Deans, Z.C. Validation of the Oncomine™ focus panel for next-generation sequencing of clinical tumour samples. Virchows Arch. 2018, 473, 489–503. [Google Scholar] [CrossRef]
- Suh, J.H.; Johnson, A.; Albacker, L.; Wang, K.; Chmielecki, J.; Frampton, G.; Gay, L.; Elvin, J.A.; Vergilio, J.A.; Ali, S.; et al. Comprehensive Genomic Profiling Facilitates Implementation of the National Comprehensive Cancer Network Guidelines for Lung Cancer Biomarker Testing and Identifies Patients Who May Benefit From Enrollment in Mechanism-Driven Clinical Trials. Oncologist 2016, 21, 684–691. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).