Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis


 , , ,
, , ,  ,
,  ,
,  ,
, 

Abstract
1. Introduction
2. Results
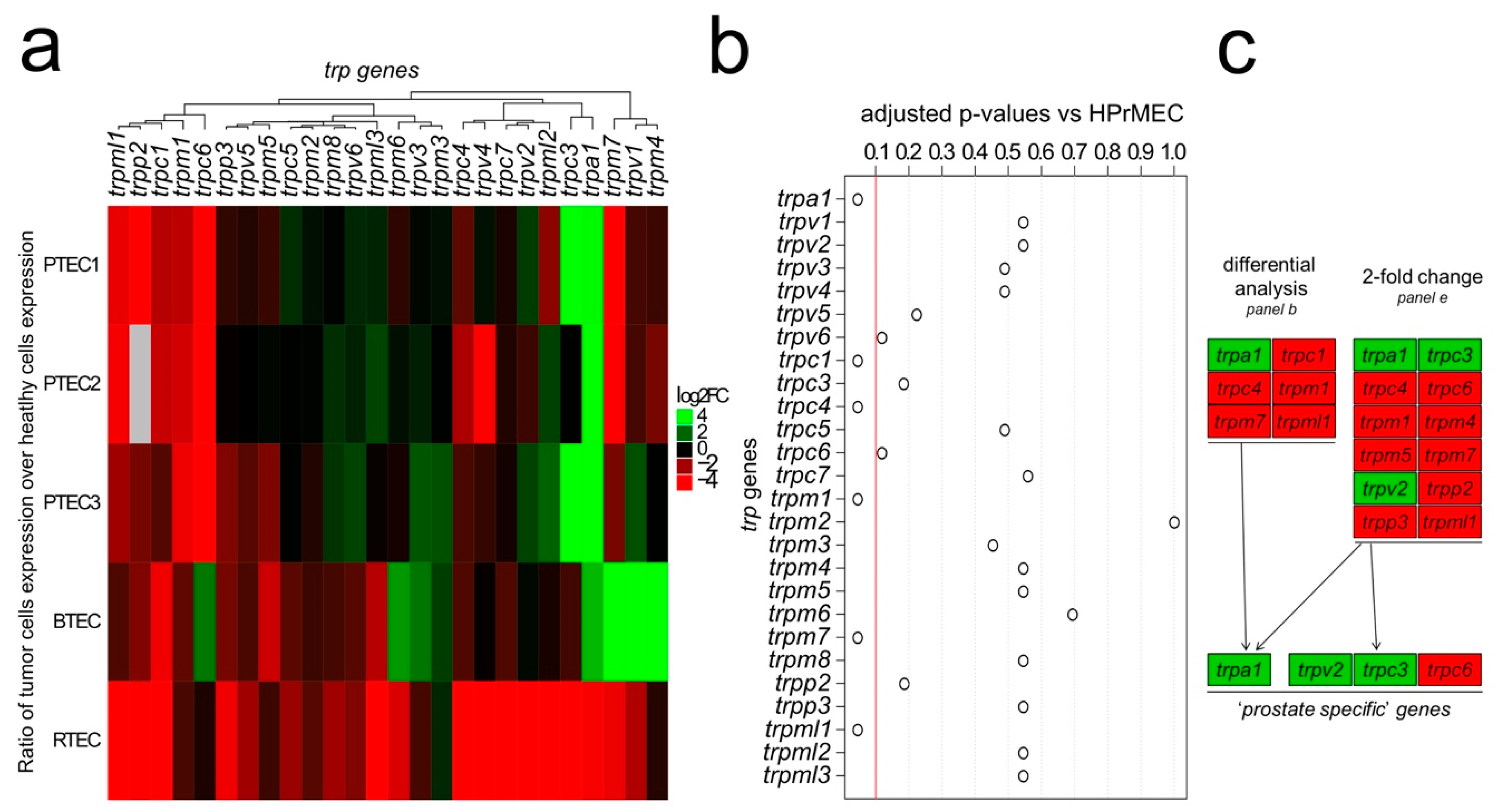
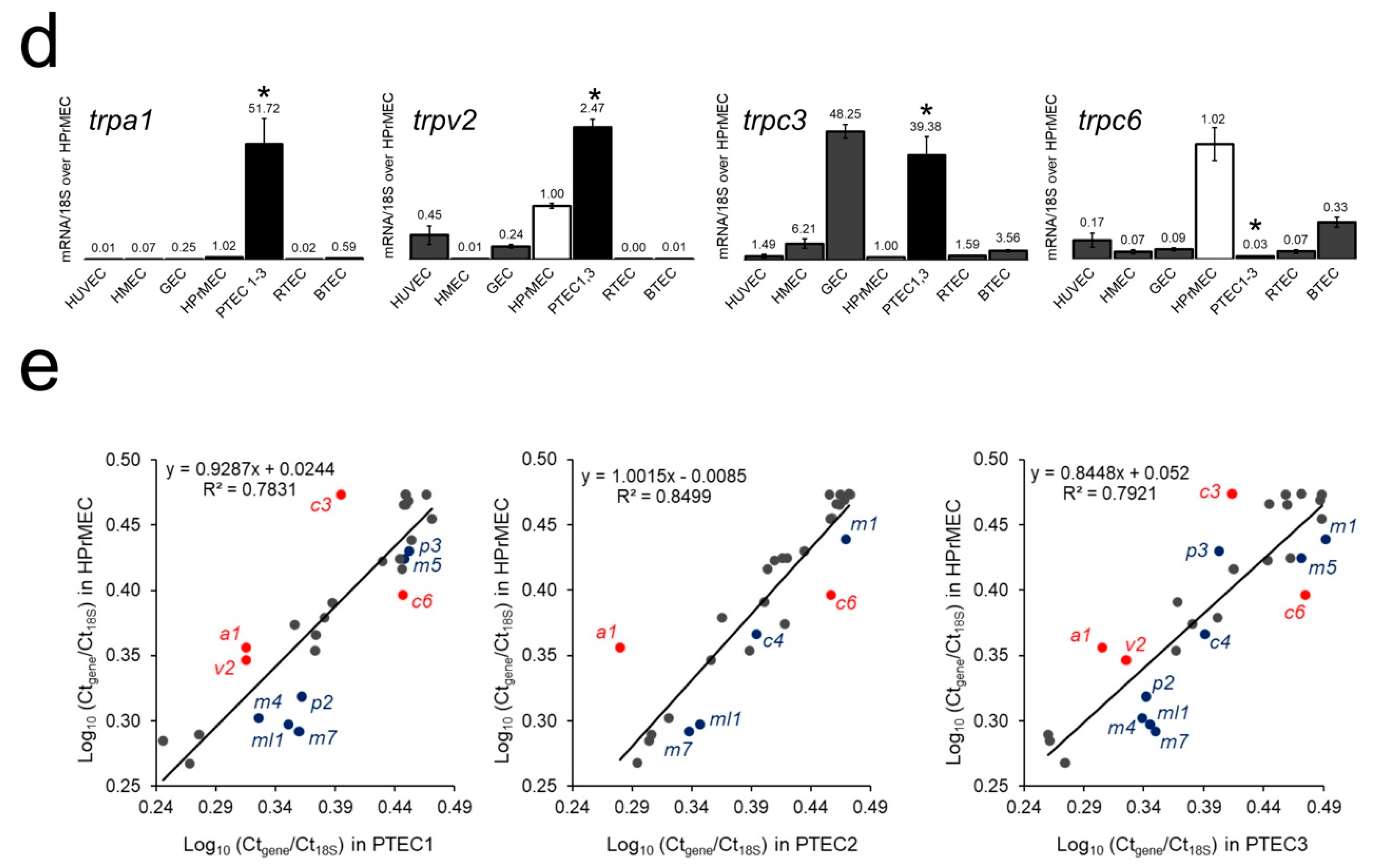
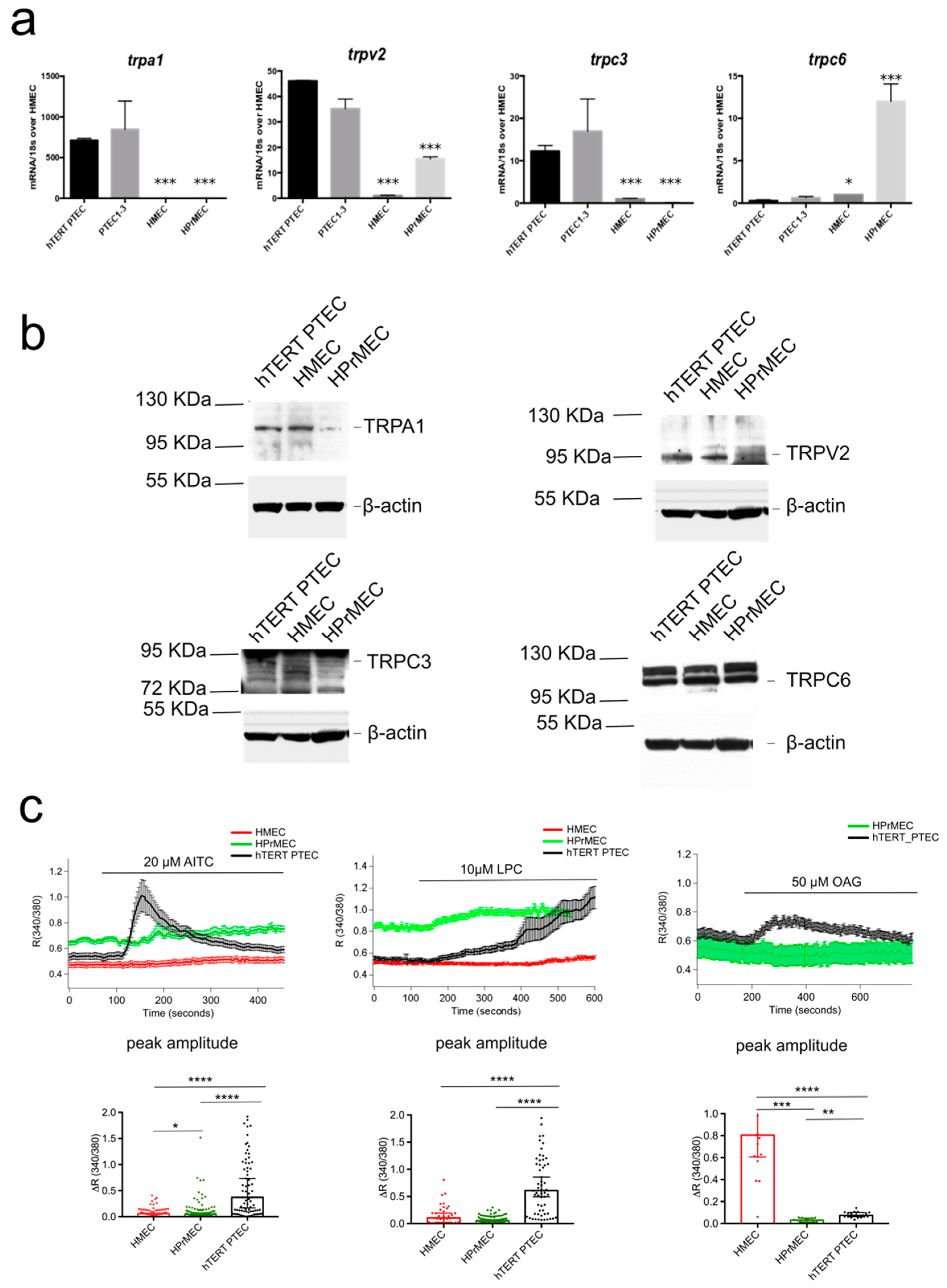
2.1. Identification of the trp Expression Signature Associated with PTEC
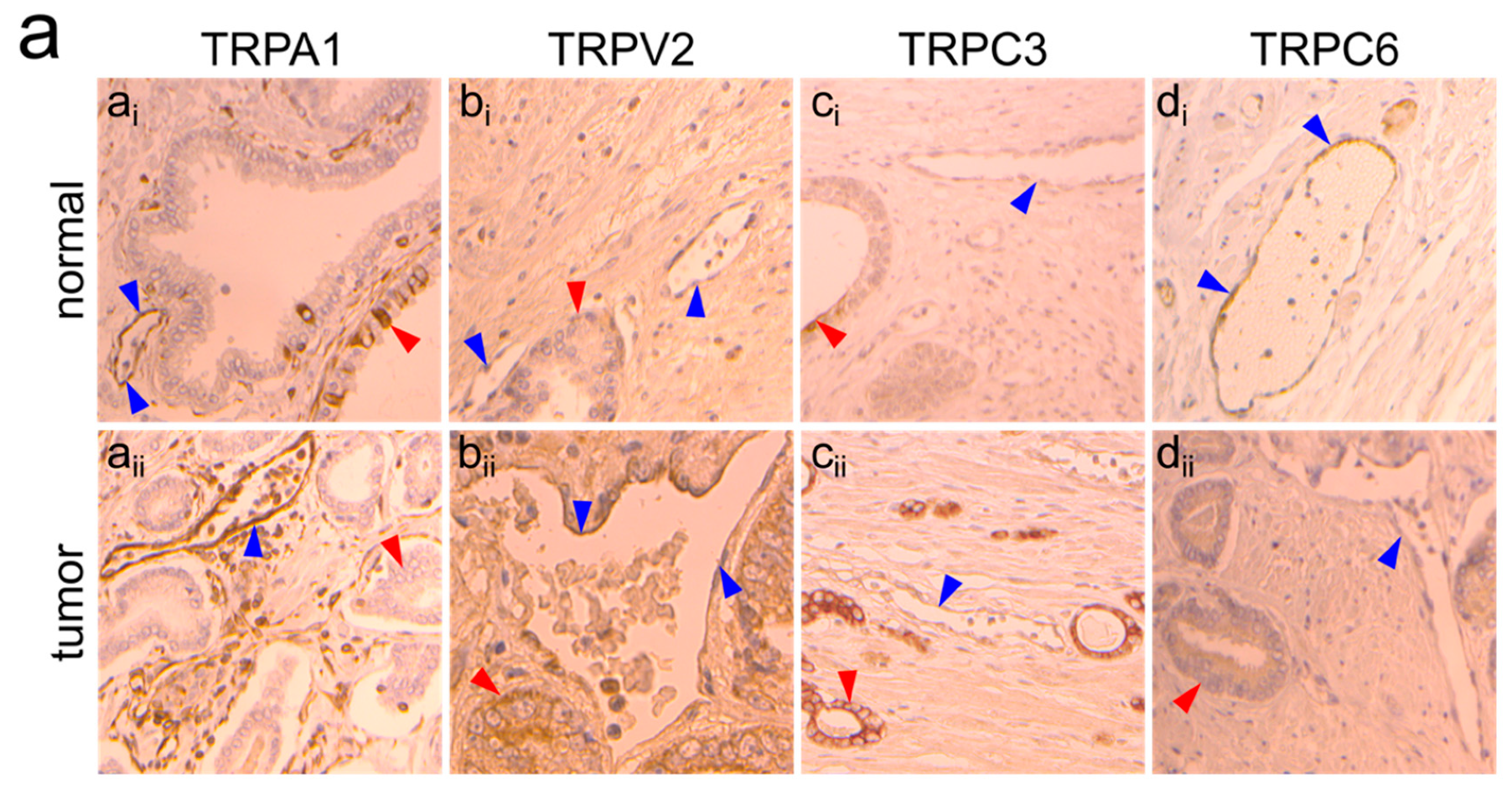
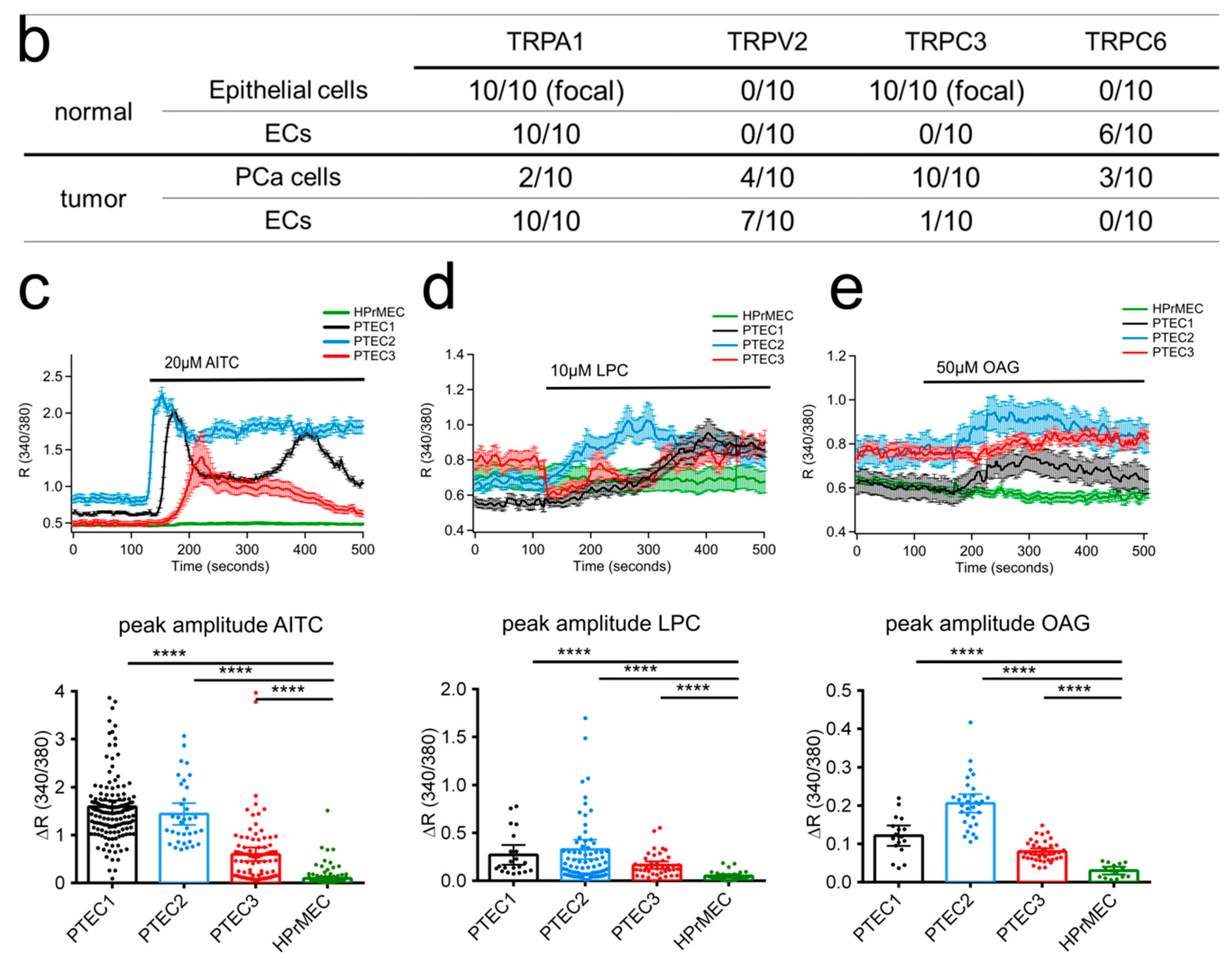
2.2. TRPA1, TRPV2, TRPC3 and TRPC6 Expression in PCa Patients
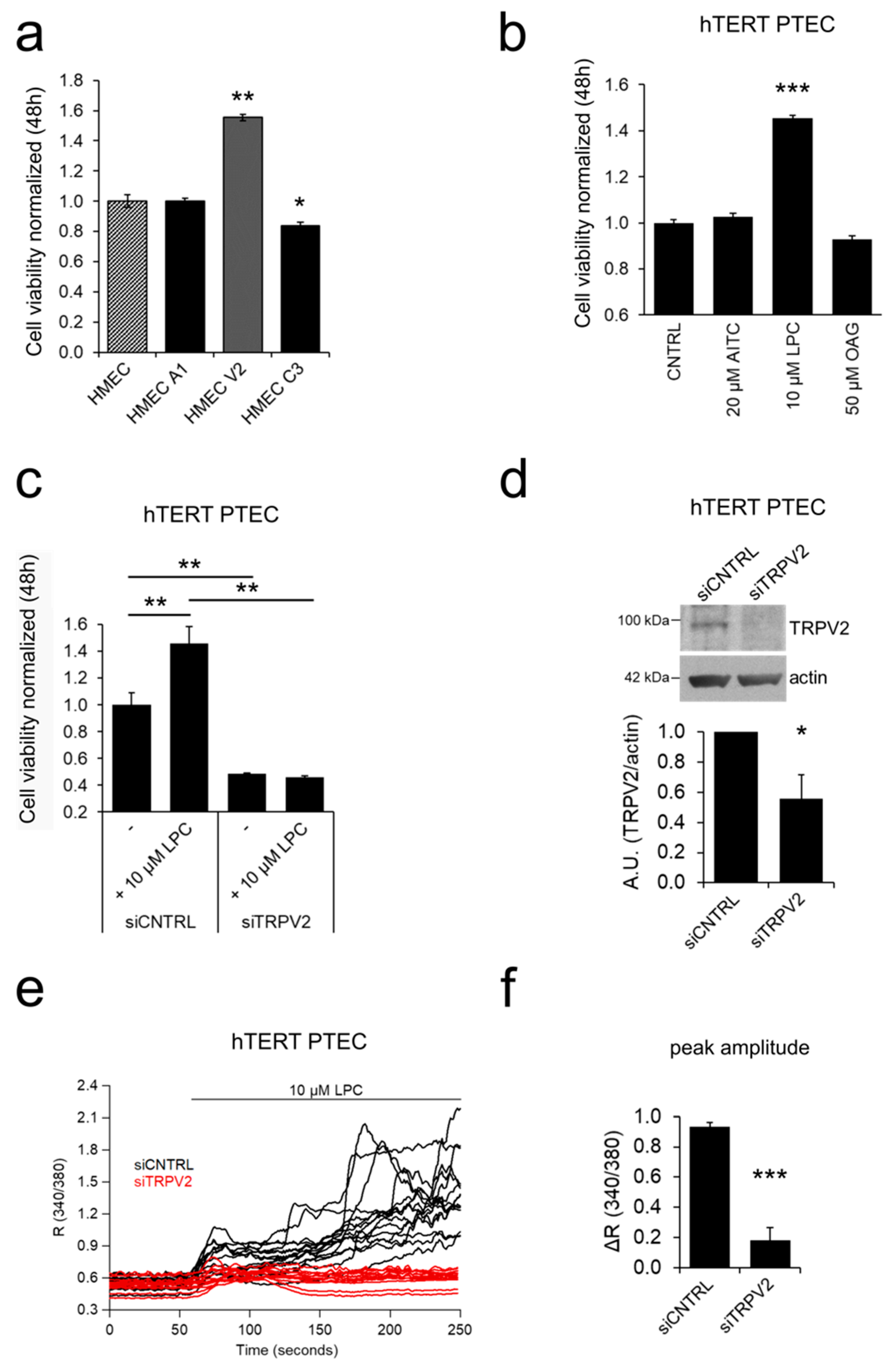
2.3. ‘Prostate-Associated’ TRPV2 Enhances EC Viability
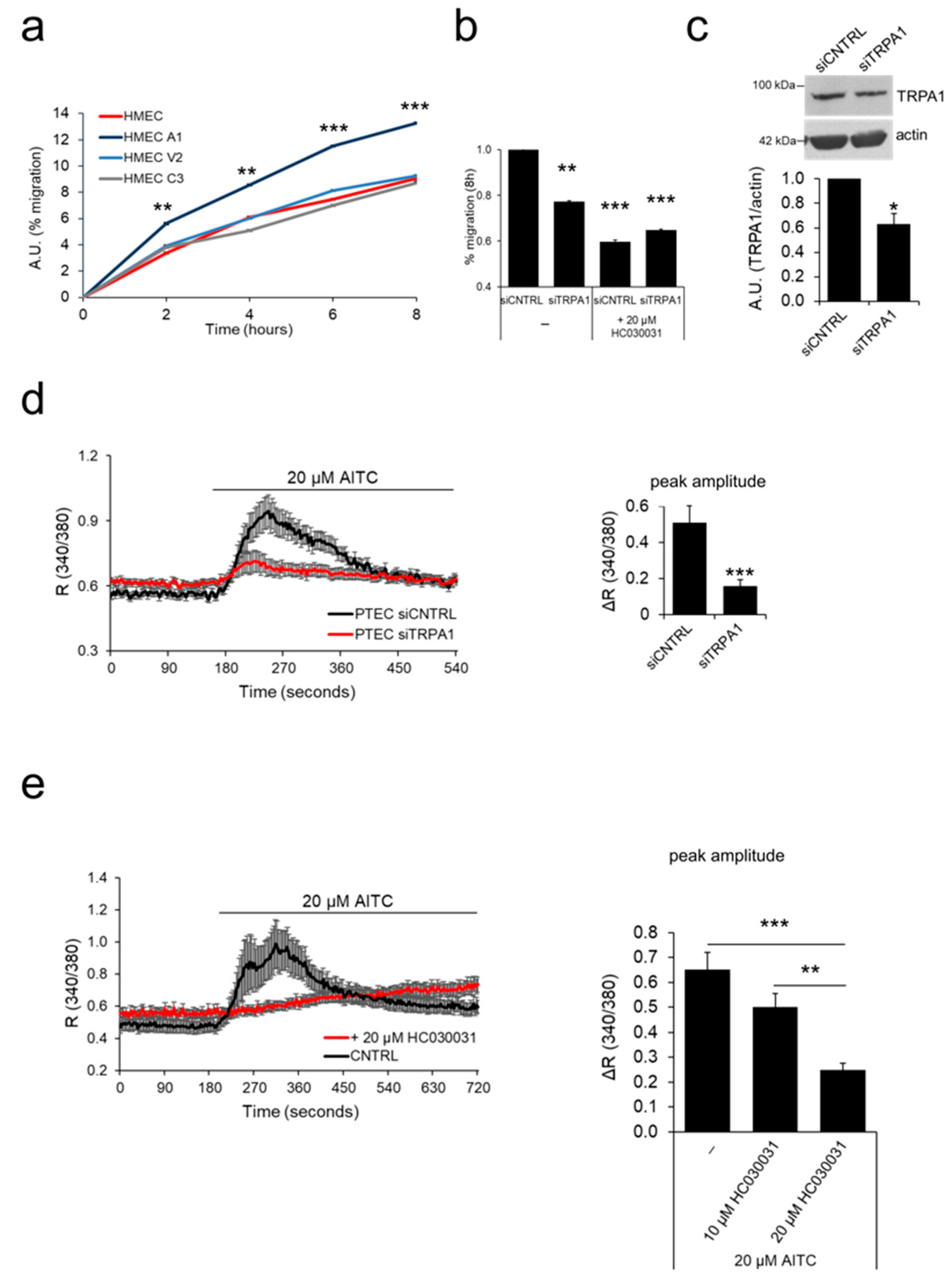
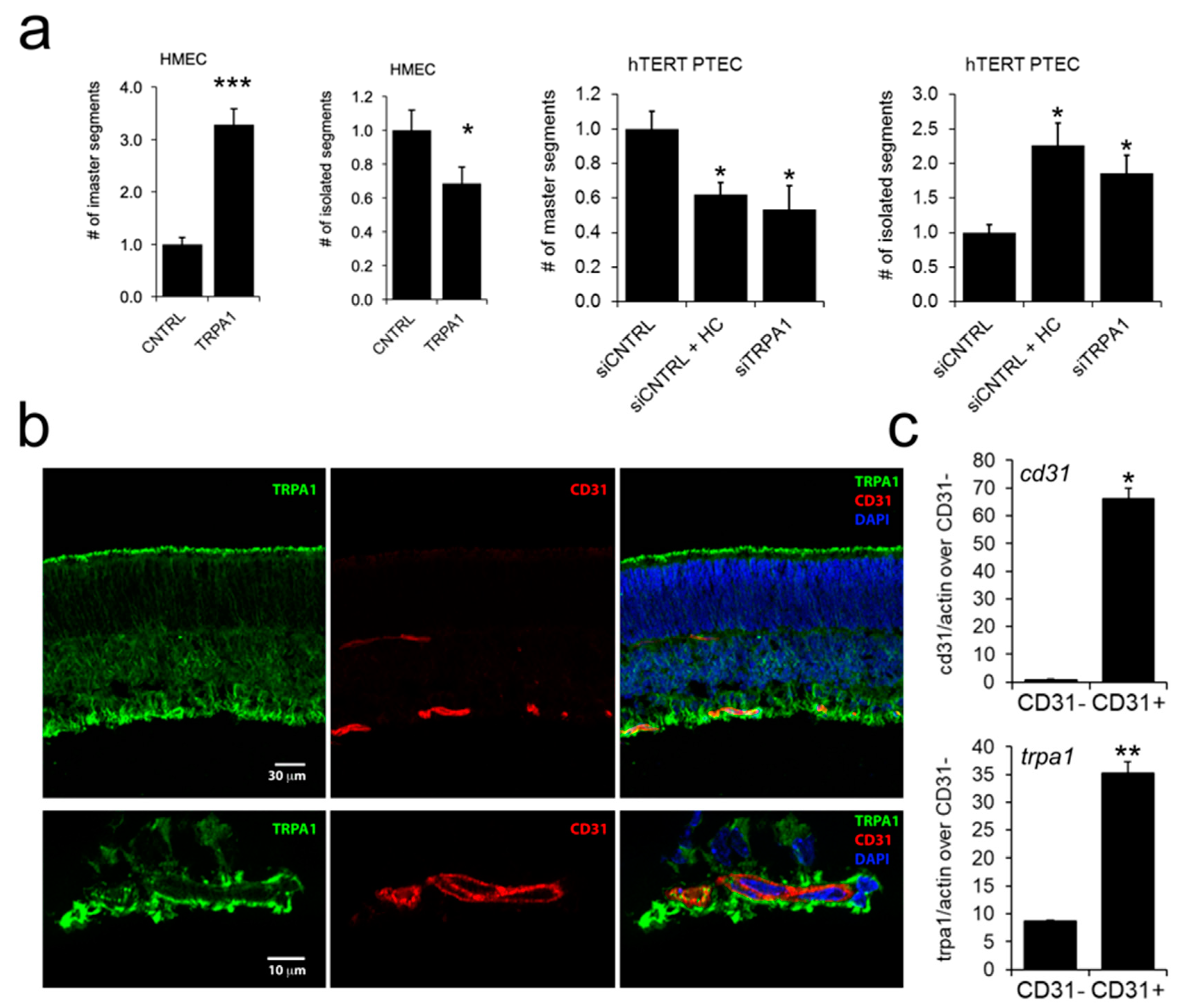
2.4. ‘Prostate-Associated’ TRPA1 Expression Promotes EC Migration
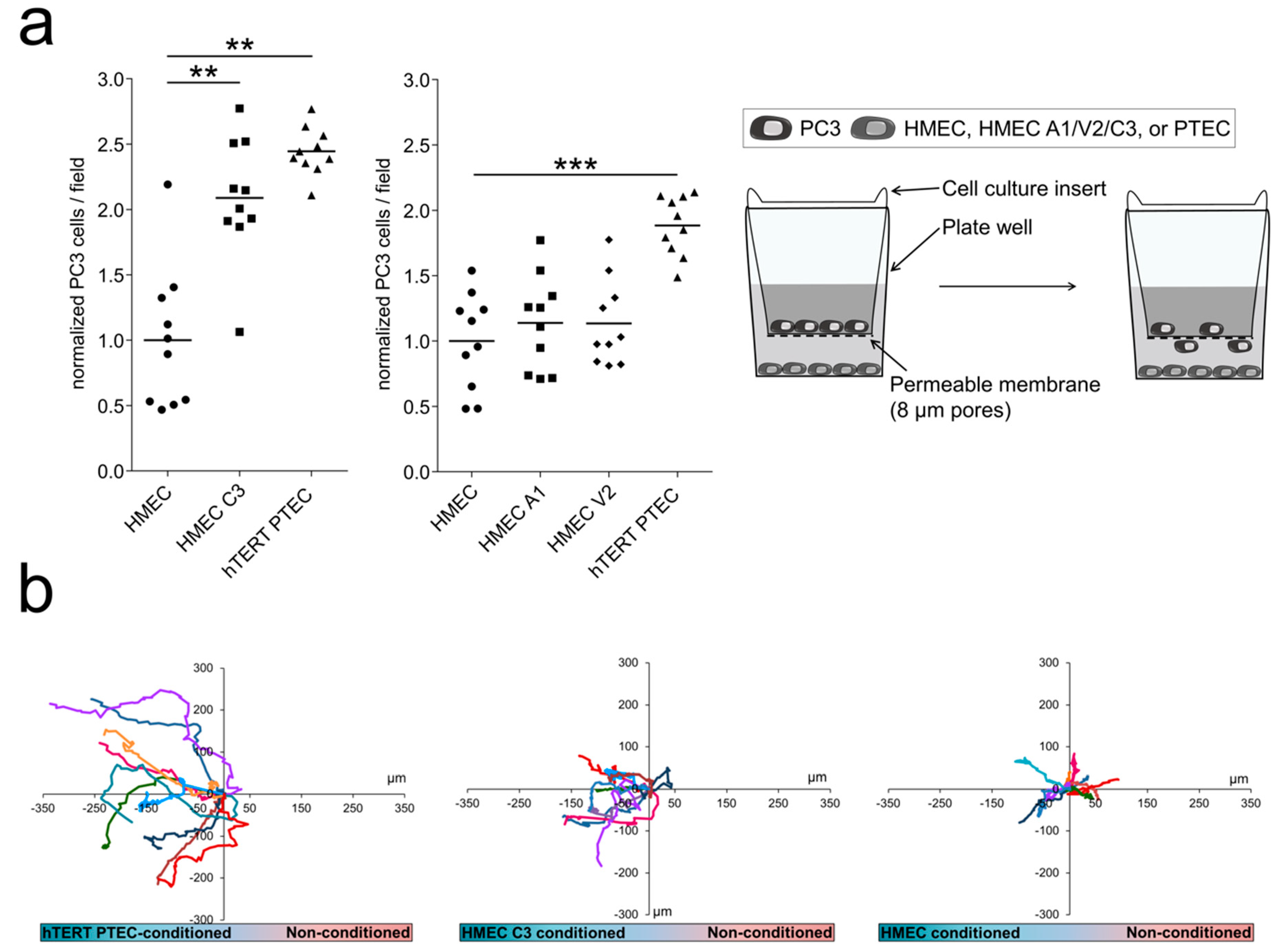
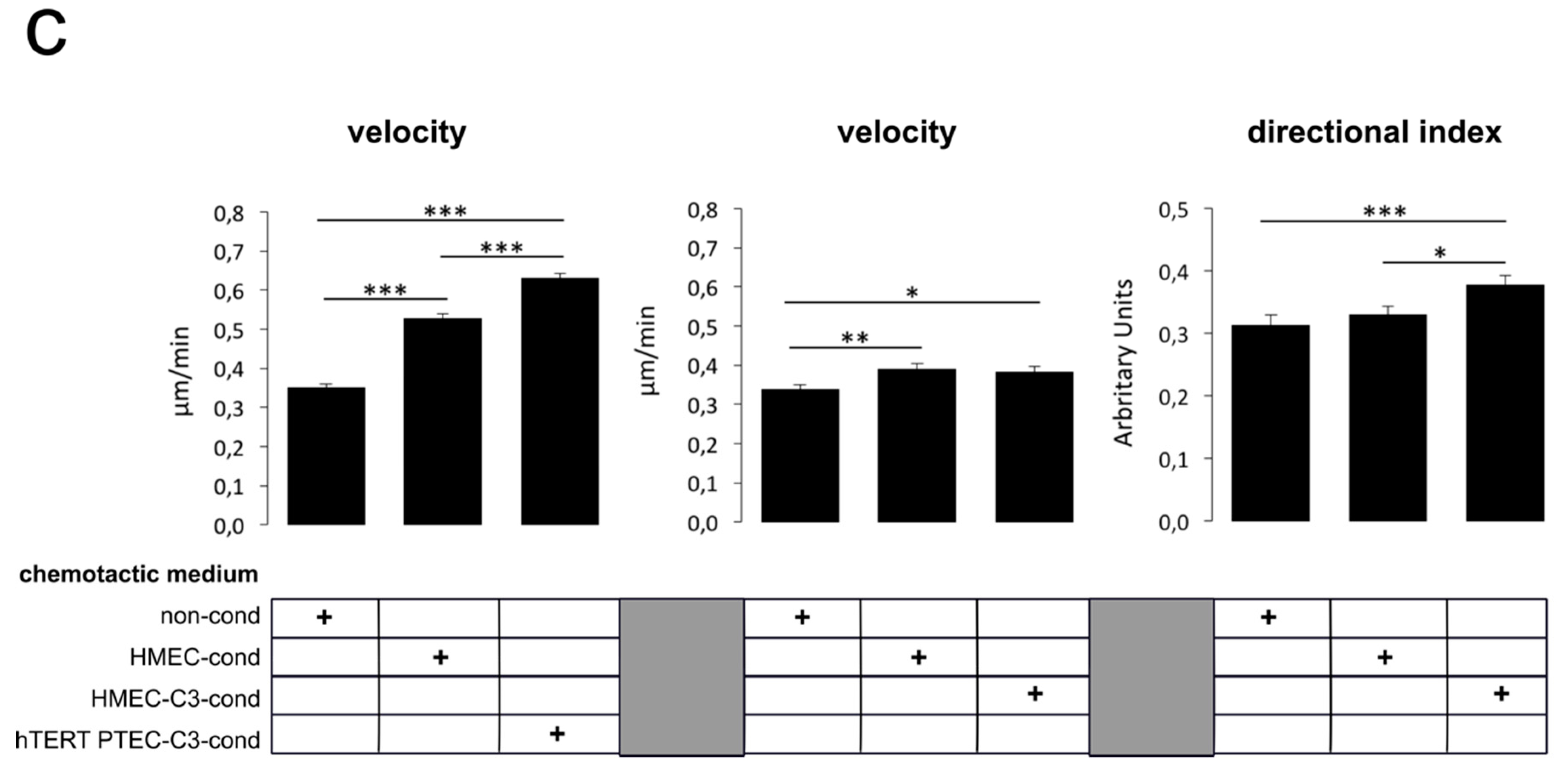
2.5. ‘Prostate-Associated’ TRPC3 Promotes PCa Cell Attraction
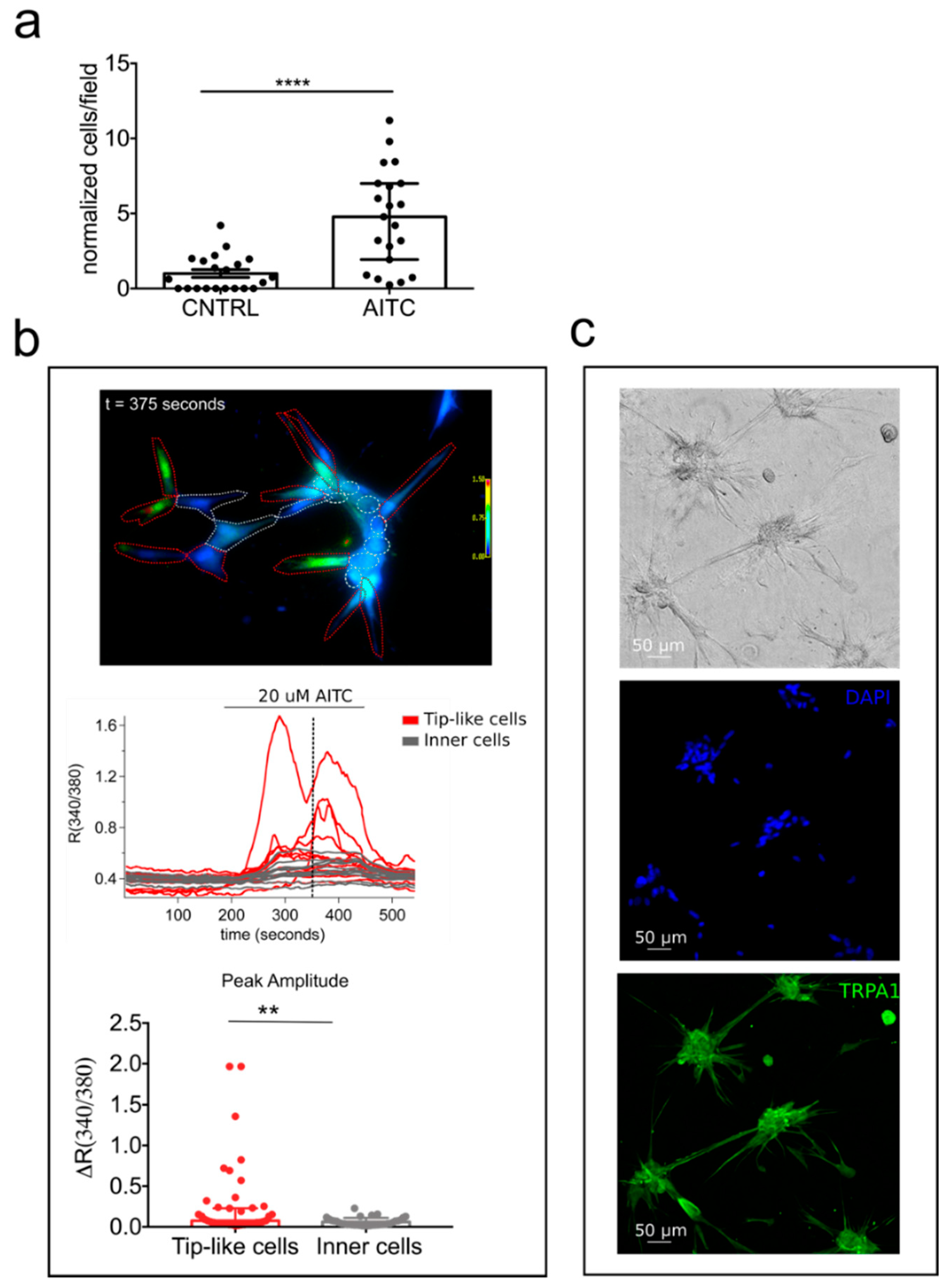
2.6. TRPA1 Promotes Vascular Network Formation and Angiogenic Sprouting both In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Drugs
4.2. Cell Cultures and Transfection
4.3. Generation of Immortalized PTEC
4.4. Flow Cytometry
4.5. In Vivo Angiogenesis
4.6. Isolation of CD31 Positive Cells from Retinas
4.7. Patients and Tissues
4.8. Immunohistochemistry
4.9. Total RNA Extraction and Reverse Transcription
4.10. Quantitative Real-Time PCR
4.11. Western Blot
4.12. Calcium Imaging
4.13. Cell Viability and Proliferation Assay
4.14. Scratch-Wound Healing Assay
4.15. Random Migration Assay
4.16. Cell Attraction Assay
4.17. 2D Chemotaxis Assay
4.18. In Vitro Tubulogenesis
4.19. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Gkika, D. Emerging role of TRP channels in cell migration: From tumor vascularization to metastasis. Front. Physiol. 2013, 4, 311. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Stock, C. Ion channels and transporters in cancer: Pathophysiology, regulation, and clinical potential. Cancer Res. 2013, 73, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.R.; Chun, J.N.; So, I.; Kim, H.J.; Baek, S.; Jeon, J.H.; Shin, S.Y. Data-driven Analysis of TRP Channels in Cancer: Linking Variation in Gene Expression to Clinical Significance. Cancer Genom. Proteom. 2016, 13, 83–90. [Google Scholar]
- Tsavaler, L.; Shapero, M.H.; Morkowski, S.; Laus, R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 2001, 61, 3760–3769. [Google Scholar] [PubMed]
- Noyer, L.; Grolez, G.P.; Prevarskaya, N.; Gkika, D.; Lemonnier, L. TRPM8 and prostate: A cold case? Pflug. Arch. 2018. [Google Scholar] [CrossRef]
- Bidaux, G.; Flourakis, M.; Thebault, S.; Zholos, A.; Beck, B.; Gkika, D.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; et al. Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. J. Clin. Investig. 2007, 117, 1647–1657. [Google Scholar] [CrossRef]
- Monet, M.; Lehen’kyi, V.; Gackiere, F.; Firlej, V.; Vandenberghe, M.; Roudbaraki, M.; Gkika, D.; Pourtier, A.; Bidaux, G.; Slomianny, C.; et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010, 70, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 channel controls prostate cancer cell proliferation via Ca2+/NFAT-dependent pathways. Oncogene 2007, 26, 7380–7385. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.; Mischi, M.; Scheepens, W.; De la Rosette, J.J.; Wijkstra, H. Angiogenesis in prostate cancer: Onset, progression and imaging. BJU Int. 2012, 110, E794–E808. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.S.; Xia, S.; Jiang, Q.; Luo, J.; Li, L.; Yeh, S.; Chang, C. Endothelial cells enhance prostate cancer metastasis via IL-6→androgen receptor→TGF-β→MMP-9 signals. Mol. Cancer Ther. 2013, 12, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Genova, T.; Grolez, G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 inhibits endothelial cell migration via a non-channel function by trapping the small GTPase Rap1. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef]
- Bernardini, M.; Fiorio Pla, A.; Prevarskaya, N.; Gkika, D. Human transient receptor potential (TRP) channel expression profiling in carcinogenesis. Int. J. Dev. Biol. 2015, 59, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Cappelli, H.C.; Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Thodeti, C.K. TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 2016, 7, 25849–25861. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Brossa, A.; Bernardini, M.; Genova, T.; Grolez, G.; Villers, A.; Leroy, X.; Prevarskaya, N.; Gkika, D.; Bussolati, B. Differential sensitivity of prostate tumor derived endothelial cells to sorafenib and sunitinib. BMC Cancer 2014, 14, 939. [Google Scholar] [CrossRef]
- Grange, C.; Bussolati, B.; Bruno, S.; Fonsato, V.; Sapino, A.; Camussi, G. Isolation and characterization of human breast tumor-derived endothelial cells. Oncol. Rep. 2006, 15, 381–386. [Google Scholar] [CrossRef][Green Version]
- Bussolati, B.; Deambrosis, I.; Russo, S.; Deregibus, M.C.; Camussi, G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003, 17, 1159–1161. [Google Scholar] [CrossRef]
- Eilken, H.M.; Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010, 22, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Pan, J.; Yu, L.; Liu, H.; Shu, X.; Sun, L.; Lou, J.; Yang, Z.; Ran, Y. Tumor endothelial cells promote metastasis and cancer stem cell-like phenotype through elevated Epiregulin in esophageal cancer. Am. J. Cancer Res. 2016, 6, 2277–2288. [Google Scholar] [PubMed]
- Ko, J.H.; Ko, E.A.; Gu, W.; Lim, I.; Bang, H.; Zhou, T. Expression profiling of ion channel genes predicts clinical outcome in breast cancer. Mol. Cancer 2013, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Gu, W.; Lim, I.; Bang, H.; Ko, E.A.; Zhou, T. Ion channel gene expression in lung adenocarcinoma: Potential role in prognosis and diagnosis. PLoS ONE 2014, 9, e86569. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Gurguis, C.I.; Gu, W.; Ko, E.A.; Lim, I.; Bang, H.; Zhou, T.; Ko, J.H. Ion channel gene expression predicts survival in glioma patients. Sci. Rep. 2015, 5, 11593. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnino, A.; Pilarsky, C.; Tawfik, D.; Sebens, S.; Trauzold, A.; Novak, I.; Schwab, A.; Kalthoff, H. In silico analysis of the transportome in human pancreatic ductal adenocarcinoma. Eur. Biophys. J. 2016, 45, 749–763. [Google Scholar] [CrossRef]
- Fantozzi, I.; Zhang, S.; Platoshyn, O.; Remillard, C.V.; Cowling, R.T.; Yuan, J.X. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1233–L1245. [Google Scholar] [CrossRef]
- Muraki, K.; Shigekawa, M.; Imaizumi, Y. A New Insight into the Function of TRPV2 in Circulatory Organs. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Laragione, T.; Cheng, K.F.; Tanner, M.R.; He, M.; Beeton, C.; Al-Abed, Y.; Gulko, P.S. The cation channel Trpv2 is a new suppressor of arthritis severity, joint damage, and synovial fibroblast invasion. Clin. Immunol. 2015, 158, 183–192. [Google Scholar] [CrossRef]
- Caprodossi, S.; Lucciarini, R.; Amantini, C.; Nabissi, M.; Canesin, G.; Ballarini, P.; Di Spilimbergo, A.; Cardarelli, M.A.; Servi, L.; Mammana, G.; et al. Transient Receptor Potential Vanilloid Type 2 (TRPV2) Expression in Normal Urothelium and in Urothelial Carcinoma of Human Bladder: Correlation with the Pathologic Stage. Eur. Urol. 2008, 54, 612–620. [Google Scholar] [CrossRef]
- Monet, M.; Gkika, D.; Lehen’kyi, V.; Pourtier, A.; Vanden Abeele, F.; Bidaux, G.; Juvin, V.; Rassendren, F.; Humez, S.; Prevarskaya, N. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim. Biophys. Acta 2009, 1793, 528–539. [Google Scholar] [CrossRef]
- Oulidi, A.; Bokhobza, A.; Gkika, D.; Vanden Abeele, F.; Lehen’kyi, V.; Ouafik, L.; Mauroy, B.; Prevarskaya, N. TRPV2 Mediates Adrenomedullin Stimulation of Prostate and Urothelial Cancer Cell Adhesion, Migration and Invasion. PLoS ONE 2013, 8, e64885. [Google Scholar] [CrossRef] [PubMed]
- Benyahia, Z.; Dussault, N.; Cayol, M.; Sigaud, R.; Berenguer-Daize, C.; Delfino, C.; Tounsi, A.; Garcia, S.; Martin, P.M.; Mabrouk, K.; et al. Stromal fibroblasts present in breast carcinomas promote tumor growth and angiogenesis through adrenomedullin secretion. Oncotarget 2017, 8, 15744–15762. [Google Scholar] [CrossRef] [PubMed]
- Linkous, A.G.; Yazlovitskaya, E.M.; Hallahan, D.E. Cytosolic phospholipase A2 and lysophospholipids in tumor angiogenesis. J. Natl. Cancer Inst. 2010, 102, 1398–1412. [Google Scholar] [CrossRef]
- Ochoa-Callejero, L.; Pozo-Rodrigalvarez, A.; Martinez-Murillo, R.; Martinez, A. Lack of adrenomedullin in mouse endothelial cells results in defective angiogenesis, enhanced vascular permeability, less metastasis, and more brain damage. Sci. Rep. 2016, 6, 33495. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ueda, T.; Shibata, Y.; Ikegami, Y.; Saito, M.; Ishida, Y.; Ugawa, S.; Kohri, K.; Shimada, S. TRPV2 activation induces apoptotic cell death in human T24 bladder cancer cells: A potential therapeutic target for bladder cancer. Urology 2010, 76, 509. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Yu, H.; Xia, Q.; Ma, Y.; Yin, H.; Shen, Y.; Liu, X. Cross-talk mechanism between endothelial cells and hepatocellular carcinoma cells via growth factors and integrin pathway promotes tumor angiogenesis and cell migration. Oncotarget 2017, 8, 69577–69593. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.A.; Miyazawa, M.; Cordeiro, M.M.; Love, W.J.; Pinsky, M.S.; Neiva, K.G.; Spalding, A.C.; Nor, J.E. Endothelial cells enhance tumor cell invasion through a crosstalk mediated by CXC chemokine signaling. Neoplasia 2008, 10, 131–139. [Google Scholar] [CrossRef]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 2015, 1848, 2685–2702. [Google Scholar] [CrossRef]
- Yokota, Y.; Nakajima, H.; Wakayama, Y.; Muto, A.; Kawakami, K.; Fukuhara, S.; Mochizuki, N. Endothelial Ca2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. Elife 2015, 4. [Google Scholar] [CrossRef]
- Yanaga, A.; Goto, H.; Nakagawa, T.; Hikiami, H.; Shibahara, N.; Shimada, Y. Cinnamaldehyde induces endothelium-dependent and -independent vasorelaxant action on isolated rat aorta. Biol. Pharm. Bull. 2006, 29, 2415–2418. [Google Scholar] [CrossRef]
- Earley, S.; Gonzales, A.L.; Crnich, R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ. Res. 2009, 104, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Derouiche, S.; Mariot, P.; Warnier, M.; Vancauwenberghe, E.; Bidaux, G.; Gosset, P.; Mauroy, B.; Bonnal, J.L.; Slomianny, C.; Delcourt, P.; et al. Activation of TRPA1 Channel by Antibacterial Agent Triclosan Induces VEGF Secretion in Human Prostate Cancer Stromal Cells. Cancer Prev. Res. 2017, 10, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Vancauwenberghe, E.; Noyer, L.; Derouiche, S.; Lemonnier, L.; Gosset, P.; Sadofsky, L.R.; Mariot, P.; Warnier, M.; Bokhobza, A.; Slomianny, C.; et al. Activation of mutated TRPA1 ion channel by resveratrol in human prostate cancer associated fibroblasts (CAF). Mol. Carcinog. 2017, 56, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Cassoni, P.; Marrocco, T.; Bussolati, B.; Allia, E.; Munaron, L.; Sapino, A.; Bussolati, G. Oxytocin induces proliferation and migration in immortalized human dermal microvascular endothelial cells and human breast tumor-derived endothelial cells. Mol. Cancer Res. 2006, 4, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Fonsato, V.; Buttiglieri, S.; Deregibus, M.C.; Puntorieri, V.; Bussolati, B.; Camussi, G. Expression of Pax2 in human renal tumor-derived endothelial cells sustains apoptosis resistance and angiogenesis. Am. J. Pathol. 2006, 168, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Hahn, W.C.; Wei, W.; Caddle, S.D.; Beijersbergen, R.L.; Lansdorp, P.M.; Sedivy, J.M.; Weinberg, R.A. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc. Natl. Acad. Sci. USA 1998, 95, 14723–14728. [Google Scholar] [CrossRef] [PubMed]
- Gkika, D.; Lemonnier, L.; Shapovalov, G.; Gordienko, D.; Poux, C.; Bernardini, M.; Bokhobza, A.; Bidaux, G.; Degerny, C.; Verreman, K.; et al. TRP channel-associated factors are a novel protein family that regulates TRPM8 trafficking and activity. J. Cell Biol. 2015, 208, 89–107. [Google Scholar] [CrossRef]
- Fiorio Pla, A.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012, 31, 200–212. [Google Scholar] [CrossRef]
- Li, J.; Shi, Y.; Toga, A.W. Controlling False Discovery Rate in Signal Space for Transformation-Invariant Thresholding of Statistical Maps. Inf. Process. Med. Imaging 2015, 24, 125–136. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRP Channel | PCR Product Length (nt) | |
|---|---|---|
| Gene | NCBI Reference Sequence | |
| trpv1 | NM_080704.3 | 101 |
| trpv2 | NM_016113.4 | 94 |
| trpv3 | NM_001258205.1 | 118 |
| trpv4 | NM_021625.4 | 99 |
| trpv5 | NM_019841.6 | 114 |
| trpv6 | NM_018646.5 | 118 |
| trpc1 | NM_001251845.1 | 118 |
| trpc3 | NM_001130698.1 | 112 |
| trpc4 | NM_016179.2 | 90 |
| trpc5 | NM_012471.2 | 111 |
| trpc6 | NM_004621.5 | 95 |
| trpc7 | NM_020389.2 | 110 |
| trpm1 | NM_001252020.1 | 91 |
| trpm2 | NM_003307.3 | 100 |
| trpm3 | NM_020952.4 | 113 |
| trpm4 | NM_017636.3 | 93 |
| trpm5 | NM_014555.3 | 117 |
| trpm6 | NM_017662.4 | 117 |
| trpm7 | NM_017672.5 | 109 |
| trpm8 | NM_024080.4 | 113 |
| trpa1 | NM_007332.2 | 95 |
| trpp2-pkd2 | NM_000297.3 | 110 |
| trpp3-pkd2l1 | NM_016112.2 | 110 |
| trpml1-mcoln1 | NM_020533.2 | 117 |
| trpml2-mcoln2 | NM_153259.3 | 91 |
| trpml3-mcoln3 | NM_018298.10 | 96 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardini, M.; Brossa, A.; Chinigò, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers 2019, 11, 956. https://doi.org/10.3390/cancers11070956
Bernardini M, Brossa A, Chinigò G, Grolez GP, Trimaglio G, Allart L, Hulot A, Marot G, Genova T, Joshi A, et al. Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers. 2019; 11(7):956. https://doi.org/10.3390/cancers11070956
Chicago/Turabian StyleBernardini, Michela, Alessia Brossa, Giorgia Chinigò, Guillaume P. Grolez, Giulia Trimaglio, Laurent Allart, Audrey Hulot, Guillemette Marot, Tullio Genova, Aditi Joshi, and et al. 2019. "Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis" Cancers 11, no. 7: 956. https://doi.org/10.3390/cancers11070956
APA StyleBernardini, M., Brossa, A., Chinigò, G., Grolez, G. P., Trimaglio, G., Allart, L., Hulot, A., Marot, G., Genova, T., Joshi, A., Mattot, V., Fromont, G., Munaron, L., Bussolati, B., Prevarskaya, N., Fiorio Pla, A., & Gkika, D. (2019). Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers, 11(7), 956. https://doi.org/10.3390/cancers11070956