PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics



Abstract
1. Introduction
2. Phosphoinositol 3 Kinase (PI3K)
2.1. Structural Overview
2.1.1. Interactions of PI3K with Upstream Regulators
2.1.2. Interactions of PI3K with Downstream Effectors
2.1.3. The Importance of PI3K 110α in OvCa
2.1.4. Correlation of PI3K Mutations in OvCa with Standard of Care Therapy
3. Protein Kinase B PKB/AKT
3.1. Structural Overview
3.1.1. Upstream AKT Activation
3.1.2. Downstream Effectors of AKT
3.1.3. Inhibition of AKT.
3.1.4. AKT in OvCa
4. Mammalian Target of Rapamycin (mTOR)
4.1. Structural Overview
4.1.1. Upstream Activation of mTORC1
4.1.2. Downstream Effectors of mTORC1
4.1.3. Upstream Regulators of mTORC2
4.1.4. Downstream Effectors of mTORC2
4.1.5. mTOR in OvCa
5. NFκB
5.1. Structural Overview
5.1.1. Canonical Pathway
5.1.2. Non-Canonical Pathway
5.1.3. Regulation of NFκB
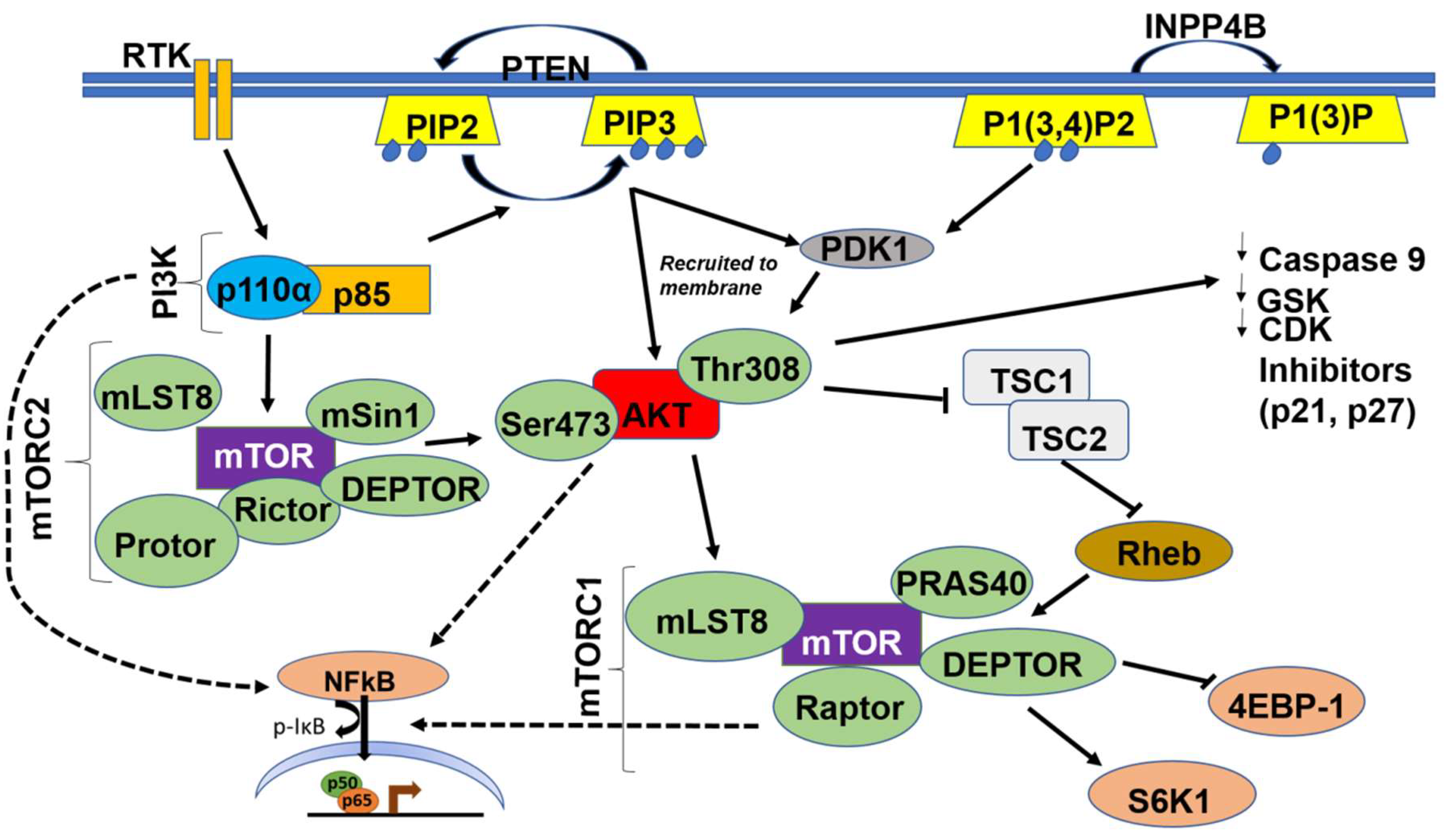
5.1.4. PI3K-AKT-mTOR-NFκB Axis
6. mTORC1 and NFκB Activation
7. Therapeutic Targeting of PI3K-AKT-mTOR-NFκB in Preclinical Models of OvCa
8. Therapeutic Targeting of PI3K-AKT-mTOR-NFκB in Clinical Trials: Opportunities and Challenges
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. Ca Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. Gepia: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Borley, J.; Wilhelm-Benartzi, C.; Brown, R.; Ghaem-Maghami, S. Does tumour biology determine surgical success in the treatment of epithelial ovarian cancer? A systematic literature review. Br. J. Cancer 2012, 107, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, W.J.; McGuire, W.P.; Brady, M.F.; Homesley, H.D.; Creasman, W.T.; Berman, M.; Ball, H.; Berek, J.S. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am. J. Obs. Gynecol. 1994, 170, 974–980. [Google Scholar] [CrossRef]
- Gasparri, M.L.; Bardhi, E.; Ruscito, I.; Papadia, A.; Farooqi, A.A.; Marchetti, C.; Bogani, G.; Ceccacci, I.; Mueller, M.D.; Benedetti Panici, P. Pi3k/akt/mtor pathway in ovarian cancer treatment: Are we on the right track? Geburtshilfe Frauenheilkd. 2017, 77, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The pi3k/akt/mtor pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. Pi3k/akt signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Greshock, J.; Colligon, T.A.; Wang, Y.; Ward, R.; Katsaros, D.; Lassus, H.; Butzow, R.; Godwin, A.K.; et al. Frequent genetic abnormalities of the pi3k/akt pathway in primary ovarian cancer predict patient outcome. Genes Chromosomes Cancer 2011, 50, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Cheaib, B.; Auguste, A.; Leary, A. The pi3k/akt/mtor pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001, 61, 7426–7429. [Google Scholar]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the pi3k-akt pathway in human cancer: Rationale and promise. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of akt1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nakayama, N.; Kurman, R.J.; Cope, L.; Pohl, G.; Samuels, Y.; Velculescu, V.E.; Wang, T.L.; Shih Ie, M. Sequence mutations and amplification of pik3ca and akt2 genes in purified ovarian serous neoplasms. Cancer Biol. 2006, 5, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals pten loss as a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Shaw, P.; Fans, I.; Rosen, B.; Risch, H.; Mitchell, C.; Sun, P.; Narod, S.A.; Kotsopoulos, J. Prognostic value of inpp4b protein immunohistochemistry in ovarian cancer. Eur. J. Gynaecol. Oncol. 2015, 36, 260–267. [Google Scholar]
- De Melo, A.C.; Paulino, E.; Garces, A.H. A review of mtor pathway inhibitors in gynecologic cancer. Oxid. Med. Cell Longev. 2017, 2017, 4809751. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. Pi3k/akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. Pi3k and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Marshall, J.D.S.; Whitecross, D.E.; Mellor, P.; Anderson, D.H. Impact of p85alpha alterations in cancer. Biomolecules 2019, 9, 29. [Google Scholar] [CrossRef]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schoneberg, J.; Ullrich, A.; Lampe, A.; Muller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef]
- Das, M.; Scappini, E.; Martin, N.P.; Wong, K.A.; Dunn, S.; Chen, Y.J.; Miller, S.L.; Domin, J.; O’Bryan, J.P. Regulation of neuron survival through an intersectin-phosphoinositide 3′-kinase c2beta-akt pathway. Mol. Cell Biol. 2007, 27, 7906–7917. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.; Bonig, H.; Kim, Y.M. Role of integrin alpha4 in drug resistance of leukemia. Front. Oncol. 2014, 4, e99. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. Pten and the pi3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.J.; Yu, L.; Backesjo, C.M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglof, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s tyrosine kinase (btk): Function, regulation, and transformation with special emphasis on the ph domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily gefs and gaps: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. Gefs and gaps: Critical elements in the control of small g proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The pi3k pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. Pi3k signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. Pi3k in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Shayesteh, L.; Lu, Y.; Kuo, W.L.; Baldocchi, R.; Godfrey, T.; Collins, C.; Pinkel, D.; Powell, B.; Mills, G.B.; Gray, J.W. Pik3ca is implicated as an oncogene in ovarian cancer. Nat. Genet. 1999, 21, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The dualistic model of ovarian carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Xu, L.; Tang, H.; Yang, Q.; Yi, X.; Fang, Y.; Zhu, Y.; Wang, Z. The role of the pten/pi3k/akt pathway on prognosis in epithelial ovarian cancer: A meta-analysis. Oncologist 2014, 19, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Musa, F.; Schneider, R. Targeting the pi3k/akt/mtor pathway in ovarian cancer. Transl. Cancer Res. 2015, 4, 97–106. [Google Scholar]
- Spangle, J.M.; Roberts, T.M.; Zhao, J.J. The emerging role of pi3k/akt-mediated epigenetic regulation in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 123–131. [Google Scholar] [CrossRef]
- Li, D.; Pan, Y.; Huang, Y.; Zhang, P.; Fang, X. Pak5 induces emt and promotes cell migration and invasion by activating the pi3k/akt pathway in ovarian cancer. Anal. Cell Pathol. 2018, 2018, e8073124. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Chou, C.H.; Wei, L.H.; Kuo, M.L.; Huang, Y.J.; Lai, K.P.; Chen, C.A.; Hsieh, C.Y. Up-regulation of interleukin-6 in human ovarian cancer cell via a gi/pi3k-akt/nf-kappab pathway by lysophosphatidic acid, an ovarian cancer-activating factor. Carcinogenesis 2005, 26, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mazzocca, A.; Giannelli, G.; Antonaci, S. Pi3k functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin. Dev. Immunol. 2011, 2011, 947858. [Google Scholar] [CrossRef] [PubMed]
- George, R.J.; Sturmoski, M.A.; Anant, S.; Houchen, C.W. Ep4 mediates pge2 dependent cell survival through the pi3 kinase/akt pathway. Prostaglandins Other Lipid Mediat. 2007, 83, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Lo, J.F.; Kuo, C.H.; Lin, J.A.; Lin, Y.M.; Chen, L.M.; Tsai, F.J.; Tsai, C.H.; Huang, C.Y.; Tang, C.H. Endothelin-1 promotes mmp-13 production and migration in human chondrosarcoma cells through fak/pi3k/akt/mtor pathways. J. Cell Physiol. 2012, 227, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Lu, P.J.; Hsiao, M.; Hsiao, C.H.; Ho, W.Y.; Cheng, P.W.; Lin, C.T.; Hong, L.Z.; Tseng, C.J. Renin activates pi3k-akt-enos signalling through the angiotensin at(1) and mas receptors to modulate central blood pressure control in the nucleus tractus solitarii. Br. J. Pharm. 2012, 166, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xu, Y. The role of lpa and yap signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 2013, 11, e31. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Follo, M.Y.; Manzoli, L.; Suh, P.G. Phosphoinositide-specific phospholipase c in health and disease. J. Lipid Res. 2015, 56, 1853–1860. [Google Scholar] [CrossRef]
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of fak-pi3k/akt pathway. Cell Biol. Int. 2014, 38, 85–91. [Google Scholar] [CrossRef]
- Le Naour, A.; Mevel, R.; Thibault, B.; Courtais, E.; Chantalat, E.; Delord, J.P.; Couderc, B.; Guillermet-Guibert, J.; Martinez, A. Effect of combined inhibition of p110 alpha pi3k isoform and stat3 pathway in ovarian cancer platinum-based resistance. Oncotarget 2018, 9, 27220–27232. [Google Scholar] [CrossRef]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis. Oncol. 2018, 2, e20. [Google Scholar] [CrossRef] [PubMed]
- Carden, C.P.; Stewart, A.; Thavasu, P.; Kipps, E.; Pope, L.; Crespo, M.; Miranda, S.; Attard, G.; Garrett, M.D.; Clarke, P.A.; et al. The association of pi3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol. Cancer 2012, 11, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Kinross, K.M.; Montgomery, K.G.; Kleinschmidt, M.; Waring, P.; Ivetac, I.; Tikoo, A.; Saad, M.; Hare, L.; Roh, V.; Mantamadiotis, T.; et al. An activating pik3ca mutation coupled with pten loss is sufficient to initiate ovarian tumorigenesis in mice. J. Clin. Investig. 2012, 122, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Brasseur, K.; Gevry, N.; Asselin, E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget 2017, 8, 4008–4042. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, G.; Getzenberg, R.H.; Veltri, R.W. The upregulation of pi3k/akt and map kinase pathways is associated with resistance of microtubule-targeting drugs in prostate cancer. J. Cell Biochem. 2015, 116, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Z.; Wang, Y.F.; Zhang, Y.; Peng, Y.M.; Liu, Y.X.; Ma, F.; Jiang, J.H.; Wang, Q.D. Cepharanthine hydrochloride reverses pglycoprotein-mediated multidrug resistance in human ovarian carcinoma a2780/taxol cells by inhibiting the pi3k/akt signaling pathway. Oncol. Rep. 2017, 38, 2558–2564. [Google Scholar] [CrossRef] [PubMed]
- Bumbaca, B.; Li, W. Taxane resistance in castration-resistant prostate cancer: Mechanisms and therapeutic strategies. Acta Pharm. Sin. B 2018, 8, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Amini-Farsani, Z.; Sangtarash, M.H.; Shamsara, M.; Teimori, H. Mir-221/222 promote chemoresistance to cisplatin in ovarian cancer cells by targeting pten/pi3k/akt signaling pathway. Cytotechnology 2018, 70, 203–213. [Google Scholar] [CrossRef]
- Wu, H.; Cao, Y.; Weng, D.; Xing, H.; Song, X.; Zhou, J.; Xu, G.; Lu, Y.; Wang, S.; Ma, D. Effect of tumor suppressor gene pten on the resistance to cisplatin in human ovarian cancer cell lines and related mechanisms. Cancer Lett. 2008, 271, 260–271. [Google Scholar] [CrossRef]
- Wu, H.; Wang, K.; Liu, W.; Hao, Q. Pten overexpression improves cisplatin-resistance of human ovarian cancer cells through upregulating krt10 expression. Biochem. Biophys. Res. Commun. 2014, 444, 141–146. [Google Scholar] [CrossRef]
- Peng, D.J.; Wang, J.; Zhou, J.Y.; Wu, G.S. Role of the akt/mtor survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010, 394, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Previs, R.A.; Sood, A.K.; Mills, G.B.; Westin, S.N. The rise of genomic profiling in ovarian cancer. Expert Rev. Mol. Diagn. 2016, 16, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Villalobos, V.; Sikic, B.I.; Plevritis, S.K. Identification of ovarian cancer driver genes by using module network integration of multi-omics data. Interface Focus 2013, 3, e20130013. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Akt/pkb signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Mundi, P.S.; Sachdev, J.; McCourt, C.; Kalinsky, K. Akt in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharm. 2016, 82, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Jin, J.; Cardiff, R.D.; Woodgett, J.R.; Muller, W.J. Activation of akt-1 (pkb-alpha) can accelerate erbb-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004, 64, 3171–3178. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; McGraw, T.E. The akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef]
- Linnerth-Petrik, N.M.; Santry, L.A.; Moorehead, R.; Jucker, M.; Wootton, S.K.; Petrik, J. Akt isoform specific effects in ovarian cancer progression. Oncotarget 2016, 7, 74820–74833. [Google Scholar] [CrossRef]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Functional specificity of akt isoforms in cancer progression. Biomol. Concepts 2011, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/pkbalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B., 3rd; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase akt2 (pkb beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for akt3/protein kinase bgamma in attainment of normal brain size. Mol. Cell Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. Mtor signaling in protein translation regulation: Implications in cancer genesis and therapeutic interventions. Mol. Biol. Int. 2014, 2014, e686984. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Wang, H.Q.; Skele, K.L.; De Rienzo, A.; Klein-Szanto, A.J.; Godwin, A.K.; Testa, J.R. Akt and mtor phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 2004, 23, 5853–5857. [Google Scholar] [CrossRef] [PubMed]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. Mtor cross-talk in cancer and potential for combination therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Woods, A.J.; Dale, T.C.; Van Der Sluijs, P.; Norman, J.C. Protein kinase b/akt acts via glycogen synthase kinase 3 to regulate recycling of alpha v beta 3 and alpha 5 beta 1 integrins. Mol. Cell Biol. 2004, 24, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of bad couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. Foxo transcription factors; regulation by akt and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/akt pathway promotes translocation of mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between akt, p53 and mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The tsc1-tsc2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4e-bp1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M. Pten tumor suppressor network in pi3k-akt pathway control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Renner, O.; Leal, J.F.; Carnero, A. Pten, more than the akt pathway. Carcinogenesis 2007, 28, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of pi(3)k-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef]
- Terakawa, N.; Kanamori, Y.; Yoshida, S. Loss of pten expression followed by akt phosphorylation is a poor prognostic factor for patients with endometrial cancer. Endocr. Relat. Cancer 2003, 10, 203–208. [Google Scholar] [CrossRef]
- Tachibana, M.; Shibakita, M.; Ohno, S.; Kinugasa, S.; Yoshimura, H.; Ueda, S.; Fujii, T.; Rahman, M.A.; Dhar, D.K.; Nagasue, N. Expression and prognostic significance of pten product protein in patients with esophageal squamous cell carcinoma. Cancer 2002, 94, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Leupin, N.; Cenni, B.; Novak, U.; Hugli, B.; Graber, H.U.; Tobler, A.; Fey, M.F. Disparate expression of the pten gene: A novel finding in b-cell chronic lymphocytic leukaemia (b-cll). Br. J. Haematol. 2003, 121, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.W.; Eom, J.I.; Maeng, H.Y.; Lee, S.T.; Hahn, J.S.; Ko, Y.W.; Min, Y.H. Phosphatase and tensin homologue phosphorylation in the c-terminal regulatory domain is frequently observed in acute myeloid leukaemia and associated with poor clinical outcome. Br. J. Haematol. 2003, 122, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Ip, L.R.; Poulogiannis, G.; Viciano, F.C.; Sasaki, J.; Kofuji, S.; Spanswick, V.J.; Hochhauser, D.; Hartley, J.A.; Sasaki, T.; Gewinner, C.A. Loss of inpp4b causes a DNA repair defect through loss of brca1, atm and atr and can be targeted with parp inhibitor treatment. Oncotarget 2015, 6, 10548–10562. [Google Scholar] [CrossRef] [PubMed]
- Fedele, C.G.; Ooms, L.M.; Ho, M.; Vieusseux, J.; O’Toole, S.A.; Millar, E.K.; Lopez-Knowles, E.; Sriratana, A.; Gurung, R.; Baglietto, L.; et al. Inositol polyphosphate 4-phosphatase ii regulates pi3k/akt signaling and is lost in human basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 22231–22236. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, Z.; Qi, M.; Wang, X.; Zhang, W.; Chen, C.; Liu, J.; Zhao, W. Inpp4b restrains cell proliferation and metastasis via regulation of the pi3k/akt/sgk pathway. J. Cell Mol. Med. 2018, 22, 2935–2943. [Google Scholar] [CrossRef]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that inositol polyphosphate 4-phosphatase type ii is a tumor suppressor that inhibits pi3k signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef]
- Hanrahan, A.J.; Schultz, N.; Westfal, M.L.; Sakr, R.A.; Giri, D.D.; Scarperi, S.; Janakiraman, M.; Olvera, N.; Stevens, E.V.; She, Q.B.; et al. Genomic complexity and akt dependence in serous ovarian cancer. Cancer Discov. 2012, 2, 56–67. [Google Scholar] [CrossRef]
- Hahne, J.C.; Honig, A.; Meyer, S.R.; Gambaryan, S.; Walter, U.; Wischhusen, J.; Haussler, S.F.; Segerer, S.E.; Fujita, N.; Dietl, J.; et al. Downregulation of akt reverses platinum resistance of human ovarian cancers in vitro. Oncol. Rep. 2012, 28, 2023–2028. [Google Scholar] [CrossRef]
- Hahne, J.C.; Meyer, S.R.; Gambaryan, S.; Walter, U.; Dietl, J.; Engel, J.B.; Honig, A. Immune escape of akt overexpressing ovarian cancer cells. Int. J. Oncol. 2013, 42, 1630–1635. [Google Scholar] [CrossRef]
- Kim, S.H.; Juhnn, Y.S.; Song, Y.S. Akt involvement in paclitaxel chemoresistance of human ovarian cancer cells. Ann. N Y Acad. Sci. 2007, 1095, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.M.; Platanias, L.C. The evolution of the tor pathway and its role in cancer. Oncogene 2013, 32, 3923–3932. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. Mtor kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Baretic, D.; Williams, R.L. The structural basis for mtor function. Semin. Cell Dev. Biol. 2014, 36, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the human mtor complex i and its implications for rapamycin inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Li, H.; Li, W.; Gui, T.; Yang, J.; Cao, D.; Shen, K. The pi3k/akt/mtor pathway is a potential predictor of distinct invasive and migratory capacities in human ovarian cancer cell lines. Oncotarget 2015, 6, 25520–25532. [Google Scholar] [CrossRef]
- Yaba, A.; Bianchi, V.; Borini, A.; Johnson, J. A putative mitotic checkpoint dependent on mtor function controls cell proliferation and survival in ovarian granulosa cells. Reprod. Sci. 2008, 15, 128–138. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mtor in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. Tor signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Hong, Z.; Pedersen, N.M.; Wang, L.; Torgersen, M.L.; Stenmark, H.; Raiborg, C. Ptdins3p controls mtorc1 signaling through lysosomal positioning. J. Cell Biol. 2017, 216, 4217–4233. [Google Scholar] [CrossRef]
- Yuan, H.X.; Russell, R.C.; Guan, K.L. Regulation of pik3c3/vps34 complexes by mtor in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. Mtor: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Musa, J.; Orth, M.F.; Dallmayer, M.; Baldauf, M.; Pardo, C.; Rotblat, B.; Kirchner, T.; Leprivier, G.; Grunewald, T.G. Eukaryotic initiation factor 4e-binding protein 1 (4e-bp1): A master regulator of mrna translation involved in tumorigenesis. Oncogene 2016, 35, 4675–4688. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ganesan, S.; Zheng, X.F. Yin and yang of 4e-bp1 in cancer. Cell Cycle 2016, 15, 1401–1402. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ding, M.; Van der Kwast, T.H.; Vellanki, R.N.; Foltz, W.D.; McKee, T.D.; Sonenberg, N.; Pandolfi, P.P.; Koritzinsky, M.; Wouters, B.G. The mtor targets 4e-bp1/2 restrain tumor growth and promote hypoxia tolerance in pten-driven prostate cancer. Mol. Cancer Res. 2018, 16, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, B.C.; Lam, J.C.; Young, R.M.; Houghton, P.J.; Keith, B.; Simon, M.C. Effects of 4e-bp1 expression on hypoxic cell cycle inhibition and tumor cell proliferation and survival. Cancer Biol. 2008, 7, 1441–1449. [Google Scholar] [CrossRef][Green Version]
- Braunstein, S.; Karpisheva, K.; Pola, C.; Goldberg, J.; Hochman, T.; Yee, H.; Cangiarella, J.; Arju, R.; Formenti, S.C.; Schneider, R.J. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol. Cell 2007, 28, 501–512. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. Ptdins(3,4,5)p3-dependent activation of the mtorc2 kinase complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A positive feedback loop between akt and mtorc2 via sin1 phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The tsc1-tsc2 complex is required for proper activation of mtor complex 2. Mol. Cell Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef]
- Thobe, K.; Sers, C.; Siebert, H. Unraveling the regulation of mtorc2 using logical modeling. Cell Commun. Signal. 2017, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of akt/pkb by the rictor-mtor complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Oh, W.J.; Jacinto, E. Mtor complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, D.; Kim, K.; Zhao, Y.; Zhao, Y.; Su, B. Mip1, an mekk2-interacting protein, controls mekk2 dimerization and activation. Mol. Cell Biol. 2005, 25, 5955–5964. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, D.C.; Qu, L.H.; Liao, H.Q.; Li, M.X. The role of mtor in ovarian cancer, polycystic ovary syndrome and ovarian aging. Clin. Anat. 2018, 31, 891–898. [Google Scholar] [CrossRef]
- Castellvi, J.; Garcia, A.; Rojo, F.; Ruiz-Marcellan, C.; Gil, A.; Baselga, J.; Ramon y Cajal, S. Phosphorylated 4e binding protein 1: A hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer 2006, 107, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to nf-kappab: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Zheng, C.; Yin, Q.A.; Wu, H. Structural studies of nf-kappa b signaling. Cell Res. 2011, 21, 183–195. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in nf-kappab signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Karin, M.; Delhase, M. The i kappa b kinase (ikk) and nf-kappa b: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Hunter, J.E.; Leslie, J.; Perkins, N.D. C-rel and its many roles in cancer: An old story with new twists. Br. J. Cancer 2016, 114, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The noncanonical nf-kappab pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Cao, Z.; Goeddel, D.V. Nf-kappab-inducing kinase activates ikk-alpha by phosphorylation of ser-176. Proc. Natl. Acad. Sci. USA 1998, 95, 3792–3797. [Google Scholar] [CrossRef] [PubMed]
- Uno, M.; Saitoh, Y.; Mochida, K.; Tsuruyama, E.; Kiyono, T.; Imoto, I.; Inazawa, J.; Yuasa, Y.; Kubota, T.; Yamaoka, S. Nf-kappab inducing kinase, a central signaling component of the non-canonical pathway of nf-kappab, contributes to ovarian cancer progression. PLoS ONE 2014, 9, e88347. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late nf-kappab-dependent gene expression. Bmc. Genom. 2010, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.C.; Chen, L.F. Regulation of nf-kappab action by reversible acetylation. Novartis. Found. Symp. 2004, 259, 208–222. [Google Scholar]
- Sachdev, S.; Hoffmann, A.; Hannink, M. Nuclear localization of ikappab alpha is mediated by the second ankyrin repeat: The ikappab alpha ankyrin repeats define a novel class of cis-acting nuclear import sequences. Mol. Cell Biol. 1998, 18, 2524–2534. [Google Scholar] [CrossRef]
- Baltimore, D. Nf-kappab is 25. Nat. Immunol. 2011, 12, 683–685. [Google Scholar] [CrossRef]
- Chen, L.F.; Greene, W.C. Regulation of distinct biological activities of the nf-kappab transcription factor complex by acetylation. J. Mol. Med. 2003, 81, 549–557. [Google Scholar] [CrossRef]
- Benezra, M.; Chevallier, N.; Morrison, D.J.; MacLachlan, T.K.; El-Deiry, W.S.; Licht, J.D. Brca1 augments transcription by the nf-kappab transcription factor by binding to the rel domain of the p65/rela subunit. J. Biol. Chem. 2003, 278, 26333–26341. [Google Scholar] [CrossRef]
- Harte, M.T.; Gorski, J.J.; Savage, K.I.; Purcell, J.W.; Barros, E.M.; Burn, P.M.; McFarlane, C.; Mullan, P.B.; Kennedy, R.D.; Perkins, N.D.; et al. Nf-kappab is a critical mediator of brca1-induced chemoresistance. Oncogene 2014, 33, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, N.; Leung, S.; Stark, G.R. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the nf-kappab p65/rela subunit. Mol. Cell Biol. 1999, 19, 4798–4805. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.A.; Huang, J.H.; Liao, W.S. Phosphatidylinositol 3-kinase in interleukin 1 signaling. Physical interaction with the interleukin 1 receptor and requirement in nfkappab and ap-1 activation. J. Biol. Chem. 1997, 272, 29167–29173. [Google Scholar] [CrossRef] [PubMed]
- Koul, D.; Yao, Y.; Abbruzzese, J.L.; Yung, W.K.; Reddy, S.A. Tumor suppressor mmac/pten inhibits cytokine-induced nfkappab activation without interfering with the ikappab degradation pathway. J. Biol. Chem. 2001, 276, 11402–11408. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. Nf-kappab activation by tumour necrosis factor requires the akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S., Jr. Akt stimulates the transactivation potential of the rela/p65 subunit of nf-kappa b through utilization of the ikappa b kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Stavnes, H.T.; Kleinberg, L.; Berner, A.; Hernandez, L.F.; Birrer, M.J.; Steinberg, S.M.; Davidson, B.; Kohn, E.C. Nuclear factor kappab transcription factors are coexpressed and convey a poor outcome in ovarian cancer. Cancer 2010, 116, 3276–3284. [Google Scholar] [CrossRef] [PubMed]
- Kleinberg, L.; Dong, H.P.; Holth, A.; Risberg, B.; Trope, C.G.; Nesland, J.M.; Florenes, V.A.; Davidson, B. Cleaved caspase-3 and nuclear factor-kappab p65 are prognostic factors in metastatic serous ovarian carcinoma. Hum. Pathol. 2009, 40, 795–806. [Google Scholar] [CrossRef]
- Jinawath, N.; Vasoontara, C.; Jinawath, A.; Fang, X.; Zhao, K.; Yap, K.L.; Guo, T.; Lee, C.S.; Wang, W.; Balgley, B.M.; et al. Oncoproteomic analysis reveals co-upregulation of rela and stat5 in carboplatin resistant ovarian carcinoma. PLoS ONE 2010, 5, e11198. [Google Scholar] [CrossRef]
- Hernandez, L.; Hsu, S.C.; Davidson, B.; Birrer, M.J.; Kohn, E.C.; Annunziata, C.M. Activation of nf-kappab signaling by inhibitor of nf-kappab kinase beta increases aggressiveness of ovarian cancer. Cancer Res. 2010, 70, 4005–4014. [Google Scholar] [CrossRef]
- Ozes, A.R.; Miller, D.F.; Ozes, O.N.; Fang, F.; Liu, Y.; Matei, D.; Huang, T.; Nephew, K.P. Nf-kappab-hotair axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene 2016, 35, 5350–5361. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of nf-{kappa}b is controlled by mtor and raptor in association with ikk. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Adli, M.; Baldwin, A.S. Regulation of mammalian target of rapamycin activity in pten-inactive prostate cancer cells by i kappa b kinase alpha. Cancer Res. 2007, 67, 6263–6269. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Basseres, D.S.; Baldwin, A.S. Akt-dependent activation of mtorc1 complex involves phosphorylation of mtor (mammalian target of rapamycin) by ikappab kinase alpha (ikkalpha). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. Ikk beta suppression of tsc1 links inflammation and tumor angiogenesis via the mtor pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A positive feedback loop involving egfr/akt/mtorc1 and ikk/nf-kb regulates head and neck squamous cell carcinoma proliferation. Oncotarget 2016, 7, 31892–31906. [Google Scholar]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa b to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, e120. [Google Scholar] [CrossRef]
- Ni, J.; Liu, Q.; Xie, S.; Carlson, C.; Von, T.; Vogel, K.; Riddle, S.; Benes, C.; Eck, M.; Roberts, T.; et al. Functional characterization of an isoform-selective inhibitor of pi3k-p110beta as a potential anticancer agent. Cancer Discov. 2012, 2, 425–433. [Google Scholar] [CrossRef]
- Schmit, F.; Utermark, T.; Zhang, S.; Wang, Q.; Von, T.; Roberts, T.M.; Zhao, J.J. Pi3k isoform dependence of pten-deficient tumors can be altered by the genetic context. Proc. Natl Acad Sci USA 2014, 111, 6395–6400. [Google Scholar] [CrossRef]
- Weigelt, B.; Warne, P.H.; Lambros, M.B.; Reis-Filho, J.S.; Downward, J. Pi3k pathway dependencies in endometrioid endometrial cancer cell lines. Clin. Cancer Res. 2013, 19, 3533–3544. [Google Scholar] [CrossRef]
- Berenjeno, I.M.; Guillermet-Guibert, J.; Pearce, W.; Gray, A.; Fleming, S.; Vanhaesebroeck, B. Both p110alpha and p110beta isoforms of pi3k can modulate the impact of loss-of-function of the pten tumour suppressor. Biochem. J. 2012, 442, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, M.; Jiang, N.; Zhang, Y.; Bian, X.; Wang, X.; Roberts, T.M.; Zhao, J.J.; Liu, P.; Cheng, H. Effective use of pi3k inhibitor bkm120 and parp inhibitor olaparib to treat pik3ca mutant ovarian cancer. Oncotarget 2016, 7, 13153–13166. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Guan, J.; Prior, W.W.; Edgar, K.A.; Kassees, R.; Sampath, D.; Belvin, M.; Friedman, L.S. Nuclear phospho-akt increase predicts synergy of pi3k inhibition and doxorubicin in breast and ovarian cancer. Sci. Transl. Med. 2010, 2, 48ra66. [Google Scholar] [CrossRef] [PubMed]
- Thomadaki, H.; Scorilas, A.; Tsiapalis, C.M.; Havredaki, M. The role of cordycepin in cancer treatment via induction or inhibition of apoptosis: Implication of polyadenylation in a cell type specific manner. Cancer Chemother Pharm. 2008, 61, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.Y.; Park, S.J.; Jo, E.; Hwang, I.H.; Lee, K.B.; Kim, S.W.; Kim, D.J.; Joo, J.C.; Hong, S.H.; Lee, M.G.; et al. Cordycepin induces apoptosis of human ovarian cancer cells by inhibiting ccl5-mediated akt/nf-kappab signaling pathway. Cell Death Discov. 2018, 4, e62. [Google Scholar] [CrossRef] [PubMed]
- Chefetz, I.; Holmberg, J.C.; Alvero, A.B.; Visintin, I.; Mor, G. Inhibition of aurora-a kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting nfkb pathway. Cell Cycle 2011, 10, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.; Ray, P. Cisplatin triggers cancer stem cell enrichment in platinum-resistant cells through nf-kappab-tnfalpha-pik3ca loop. J. Exp. Clin. Cancer Res. 2017, 36, e164. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Torres, C.; Gaytan-Cervantes, J.; Vazquez-Santillan, K.; Mandujano-Tinoco, E.A.; Ceballos-Cancino, G.; Garcia-Venzor, A.; Zampedri, C.; Sanchez-Maldonado, P.; Mojica-Espinosa, R.; Jimenez-Hernandez, L.E.; et al. Nf-kappab participates in the stem cell phenotype of ovarian cancer cells. Arch. Med. Res. 2017, 48, 343–351. [Google Scholar] [CrossRef]
- House, C.D.; Jordan, E.; Hernandez, L.; Ozaki, M.; James, J.M.; Kim, M.; Kruhlak, M.J.; Batchelor, E.; Elloumi, F.; Cam, M.C.; et al. Nfkappab promotes ovarian tumorigenesis via classical pathways that support proliferative cancer cells and alternative pathways that support aldh(+) cancer stem-like cells. Cancer Res. 2017, 77, 6927–6940. [Google Scholar] [CrossRef]
- Tang, S.; Xiang, T.; Huang, S.; Zhou, J.; Wang, Z.; Xie, R.; Long, H.; Zhu, B. Ovarian cancer stem-like cells differentiate into endothelial cells and participate in tumor angiogenesis through autocrine ccl5 signaling. Cancer Lett. 2016, 376, 137–147. [Google Scholar] [CrossRef]
- Jin, Z.; Niu, H.; Wang, X.; Zhang, L.; Wang, Q.; Yang, A. Preclinical study of cc223 as a potential anti-ovarian cancer agent. Oncotarget 2017, 8, 58469–58479. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting sphk1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Vadas, M.; Xia, P.; McCaughan, G.; Gamble, J. The role of sphingosine kinase 1 in cancer: Oncogene or non-oncogene addiction? Biochim. Biophys. Acta 2008, 1781, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Heo, J.H.; Park, J.Y.; Jeong, J.Y.; Cho, H.J.; Park, K.S.; Kim, S.H.; Moon, Y.W.; Kim, J.S.; An, H.J. A novel pi3k/mtor dual inhibitor, cmg002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019, 153, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Speranza, M.C.; Nowicki, M.O.; Behera, P.; Cho, C.F.; Chiocca, E.A.; Lawler, S.E. Bkm-120 (buparlisib): A phosphatidyl-inositol-3 kinase inhibitor with anti-invasive properties in glioblastoma. Sci. Rep. 2016, 6, e20189. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (byl719) in pik3ca-altered solid tumors: Results from the first-in-human study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Javle, M.; Bekaii-Saab, T.S.; Finn, R.S.; Wainberg, Z.A.; Laheru, D.A.; Weekes, C.D.; Tan, B.R.; Khan, G.N.; Zalupski, M.M.; et al. A phase 1 dose-escalation and expansion study of binimetinib (mek162), a potent and selective oral mek1/2 inhibitor. Br. J. Cancer 2017, 116, 575–583. [Google Scholar] [CrossRef]
- Mensah, F.A.; Blaize, J.P.; Bryan, L.J. Spotlight on copanlisib and its potential in the treatment of relapsed/refractory follicular lymphoma: Evidence to date. Onco. Targets 2018, 11, 4817–4827. [Google Scholar] [CrossRef]
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.; Ma, A.W.; Xu, G.X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual hdac and pi3k inhibitor cudc-907 downregulates myc and suppresses growth of myc-dependent cancers. Mol. Cancer 2017, 16, 285–299. [Google Scholar] [CrossRef]
- Caro-Vegas, C.; Bailey, A.; Bigi, R.; Damania, B.; Dittmer, D.P. Targeting mtor with mln0128 overcomes rapamycin and chemoresistant primary effusion lymphoma. MBio 2019, 10, e02871-18. [Google Scholar] [CrossRef]
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef]
- Chen, W.C.; Lai, Y.A.; Lin, Y.C.; Ma, J.W.; Huang, L.F.; Yang, N.S.; Ho, C.T.; Kuo, S.C.; Way, T.D. Curcumin suppresses doxorubicin-induced epithelial-mesenchymal transition via the inhibition of tgf-beta and pi3k/akt signaling pathways in triple-negative breast cancer cells. J. Agric. Food Chem. 2013, 61, 11817–11824. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Kim, C.G.; Lim, Y.; Shin, S.Y.; Lee, Y.H. Curcumin down-regulates the multidrug-resistance mdr1b gene by inhibiting the pi3k/akt/nf kappa b pathway. Cancer Lett. 2008, 259, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Sesarman, A.; Tefas, L.; Sylvester, B.; Licarete, E.; Rauca, V.; Luput, L.; Patras, L.; Banciu, M.; Porfire, A. Anti-angiogenic and anti-inflammatory effects of long-circulating liposomes co-encapsulating curcumin and doxorubicin on c26 murine colon cancer cells. Pharmacol. Rep. Pr. 2018, 70, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Sesarman, A.; Tefas, L.; Sylvester, B.; Licarete, E.; Rauca, V.; Luput, L.; Patras, L.; Porav, S.; Banciu, M.; Porfire, A. Co-delivery of curcumin and doxorubicin in pegylated liposomes favored the antineoplastic c26 murine colon carcinoma microenvironment. Drug Deliv. Transl. Res. 2019, 9, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Kaklamani, V.G.; Richardson, A.L.; Arteaga, C.L. Exploring biomarkers of phosphoinositide 3-kinase pathway activation in the treatment of hormone receptor positive, human epidermal growth receptor 2 negative advanced breast cancer. Oncologist 2019, 24, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting inflammation in cancer prevention and therapy. Cancer Prev. Res. 2016, 9, 895–905. [Google Scholar] [CrossRef] [PubMed]







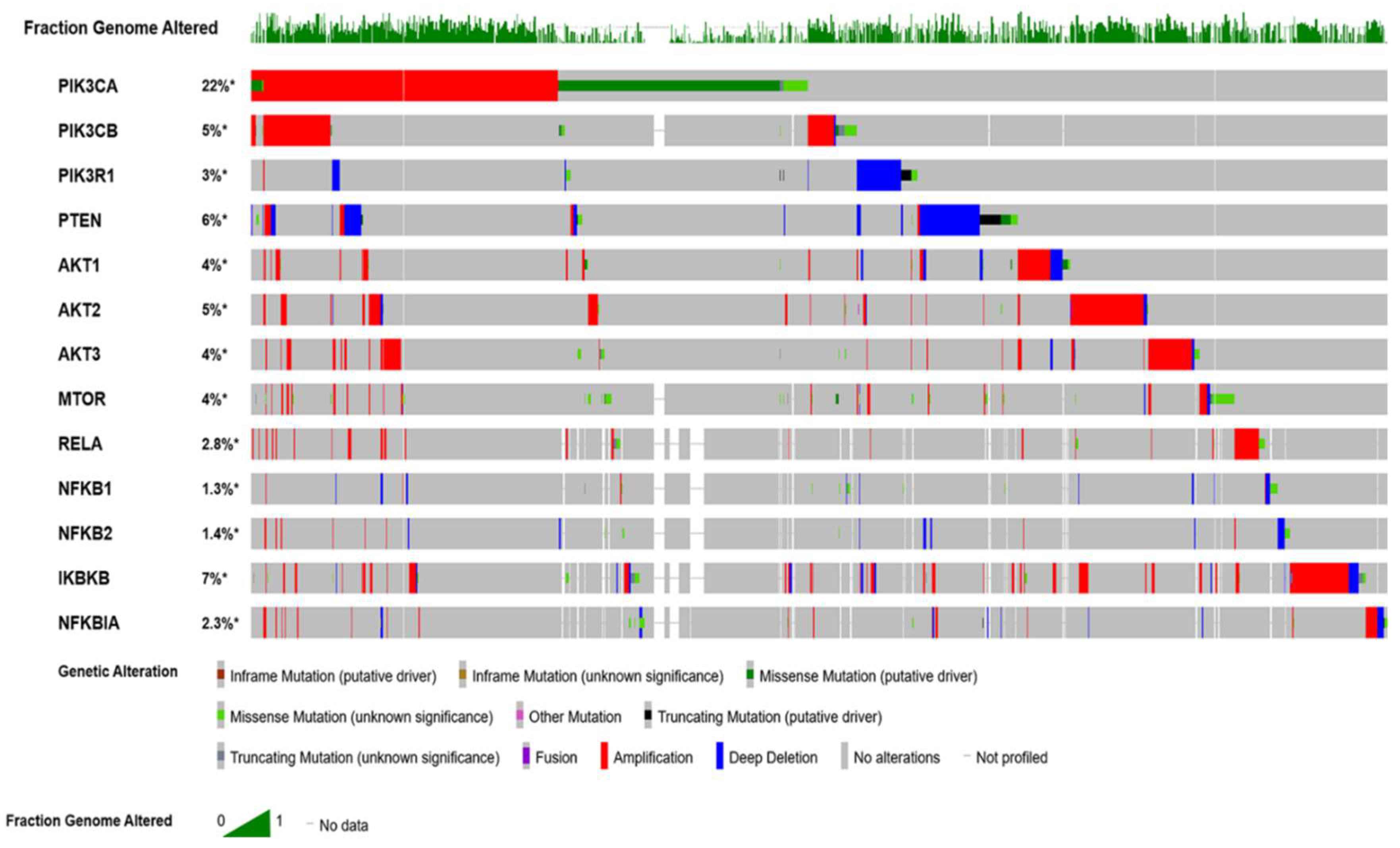{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Annotation | PI3K | PTEN | AKT |
|---|---|---|---|---|
| A2780 | Unlikely HGSC/not-specified | PIK3CA Missense p.E365K | Deletion | AKT3 Gain |
| CAOV3 | Likely HGSC/adenocarcinoma | PIK3CA amplification | Gain | AKT1/2 Gain |
| IGROV1 | Mixed subtype/hypermutated | PIK3CA Missense/non-stop p.R38C & p.1069W | p.Y155C Loss of function | |
| PIK3CB p.A686V | ||||
| MCAS | Mucinous cystadenocarcinoma | PIK3CA Missense p.H1047R | Gain | |
| OVMIU | Not-specified | PIK3CA Missense p.H1047R | ||
| OVMANA | Clear-cell | PIK3CA Missense p.E545V | ||
| OVISE | Clear-cell | PIK3CA Missense p.C420R | AKT2 Gain | |
| OC314 | Serous | PIK3CA Missense p.R108H | ||
| OC316 | Low-differentiated adenocarcinoma | PIK3CA Missense p.R108H | Heterozygous loss | AKT1 Gain |
| PIK3CG p.T309M & p.R542W | ||||
| EFO27 | Mucinous | PIK3CA Missense p.H510N | ||
| COV318 | Serous/non-specified? | PIK3CA Gain/amplification? | AKT1/2/3 Gain | |
| PIK3CB Missense p.W676R & p.V260I | ||||
| OVCAR3 | Poorly differentiated papillary adenocarcinoma/possibly HGSC | PIK3CA AMP | AKT2 amplification/Gain | |
| PIK3R1 splice variant c.e14-1 (likely loss of function) | ||||
| SKOV3 | Moderately well-differentiated adenocarcinoma/mixed subtype | PIK3CA Missense p.H1047R | Heterozygous loss | |
| TOV21G | Clear-cell adenocarcinoma/hyper-mutated | PIK3CA Missense p.H1047Y | Gain | |
| OAW42 | Non-specified | PIK3CA Missense p.H1047L | Heterozygous loss | AKT2 Gain |
| FUOV1 | Serous | AKT1/2 Gain |
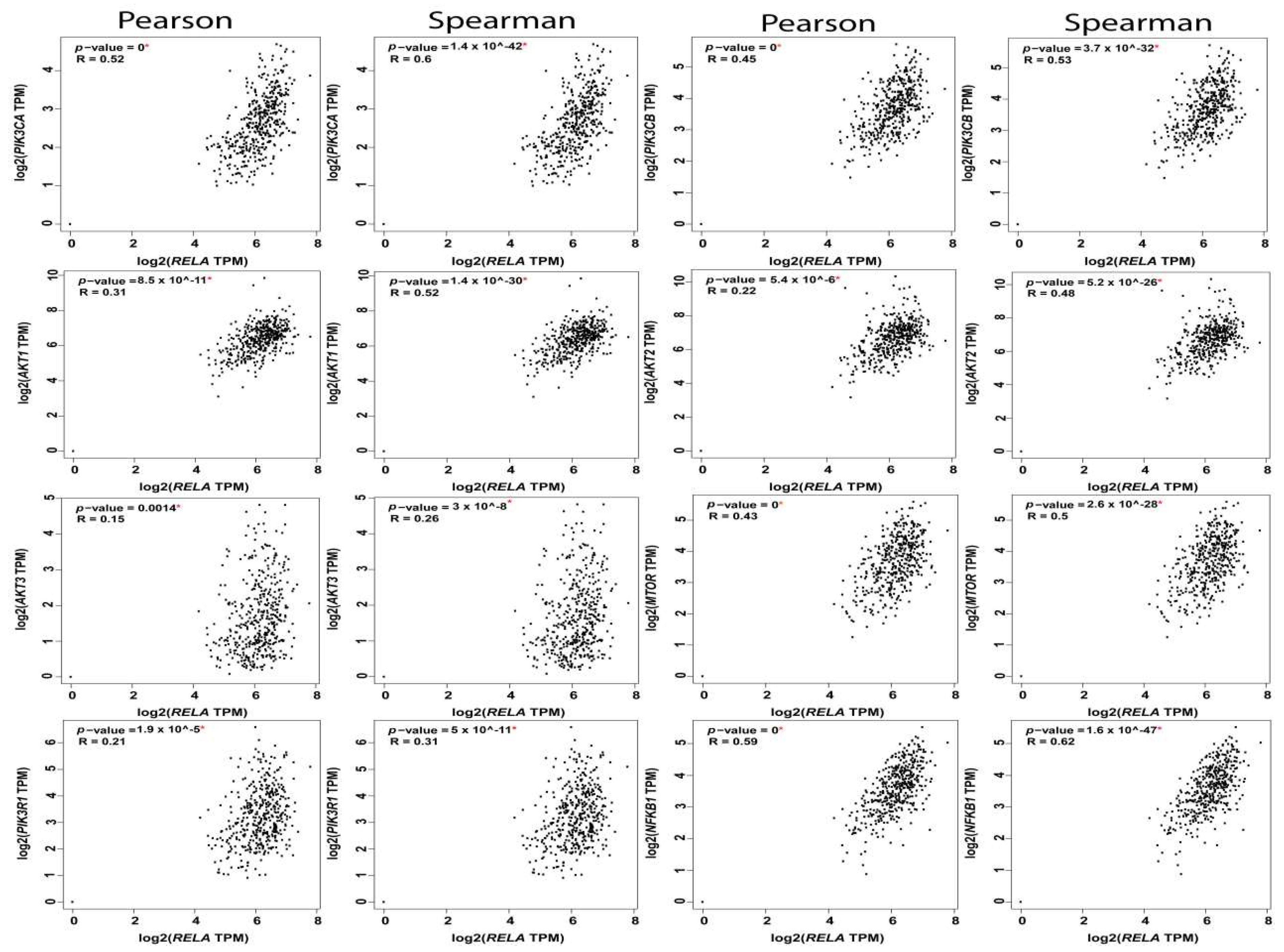
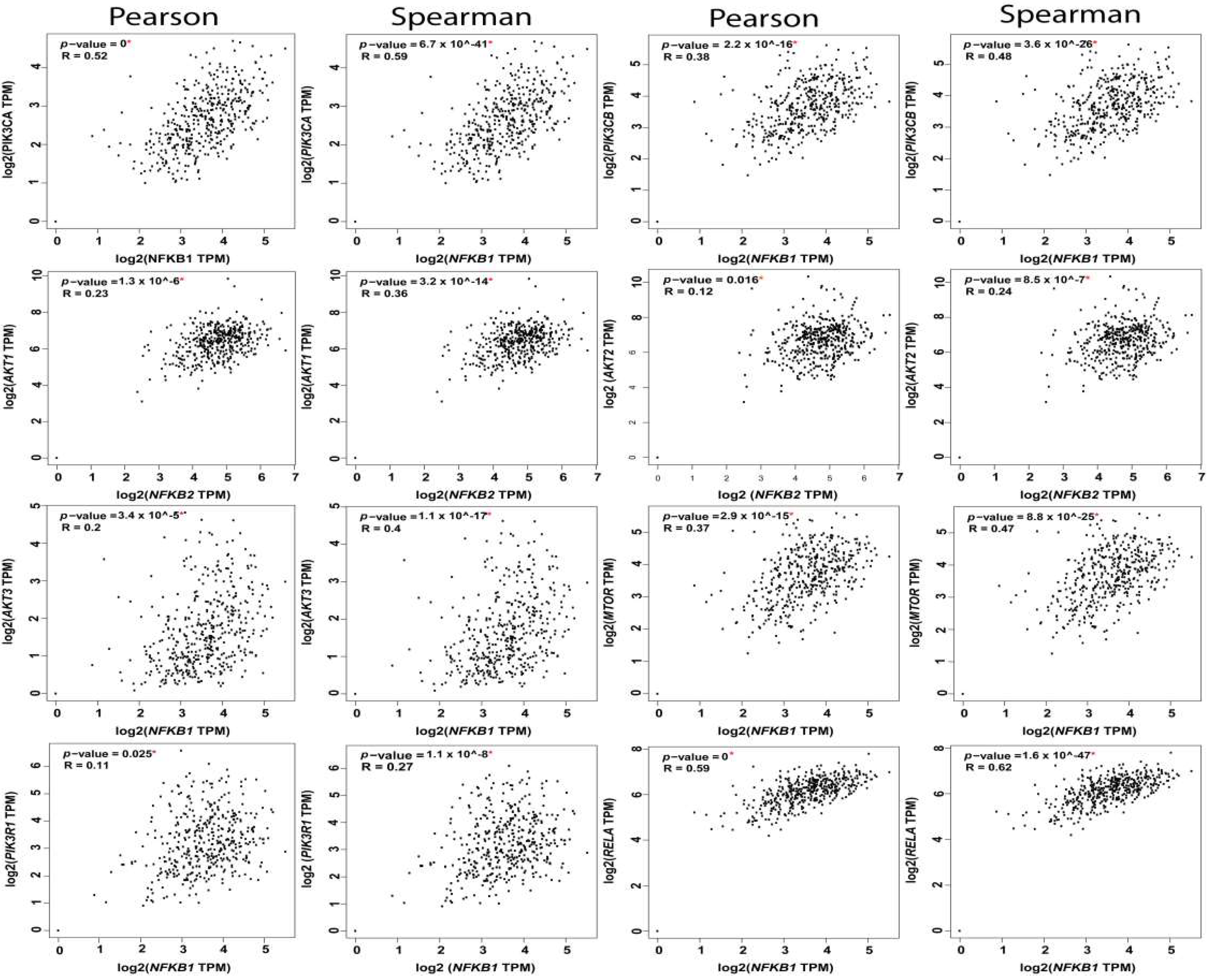
| A | B | Both | Log2 Odds Ratio | p-Value | q-Value |
|---|---|---|---|---|---|
| PIK3CA | PIK3CB | 117 | 2.813 | <0.001 | <0.001 |
| PIK3CA | AKT3 | 60 | 1.272 | <0.001 | <0.001 |
| PIK3CB | MTOR | 21 | 2.009 | <0.001 | <0.001 |
| PTEN | AKT1 | 23 | 1.814 | <0.001 | <0.001 |
| AKT1 | IKBKB | 23 | 1.659 | <0.001 | <0.001 |
| PIK3CA | PTEN | 75 | 0.918 | <0.001 | <0.001 |
| PIK3R1 | MTOR | 14 | 2.132 | <0.001 | <0.001 |
| PIK3CA | RELA | 37 | 1.343 | <0.001 | <0.001 |
| PIK3CA | MTOR | 47 | 1.109 | <0.001 | <0.001 |
| AKT3 | MTOR | 16 | 1.808 | <0.001 | <0.001 |
| AKT2 | AKT3 | 20 | 1.576 | <0.001 | <0.001 |
| PTEN | NFKBIA | 15 | 1.875 | <0.001 | <0.001 |
| PIK3R1 | IKBKB | 19 | 1.607 | <0.001 | <0.001 |
| AKT1 | NFKB2 | 8 | 2.582 | <0.001 | <0.001 |
| PTEN | MTOR | 19 | 1.484 | <0.001 | 0.001 |
| AKT2 | IKBKB | 26 | 1.247 | <0.001 | 0.001 |
| IKBKB | NFKBIA | 14 | 1.598 | <0.001 | 0.004 |
| PIK3CB | NFKB1 | 8 | 2.15 | 0.001 | 0.005 |
| PIK3R1 | AKT1 | 11 | 1.679 | 0.002 | 0.006 |
| PTEN | NFKB2 | 9 | 1.929 | 0.002 | 0.006 |
| PIK3CB | NFKB2 | 8 | 2.034 | 0.002 | 0.007 |
| PIK3CA | AKT2 | 57 | 0.707 | 0.002 | 0.008 |
| AKT3 | NFKB2 | 7 | 2.08 | 0.003 | 0.009 |
| MTOR | NFKB1 | 6 | 2.205 | 0.004 | 0.011 |
| PIK3CB | AKT1 | 14 | 1.248 | 0.005 | 0.016 |
| PIK3CB | NFKBIA | 10 | 1.474 | 0.006 | 0.019 |
| AKT2 | NFKBIA | 10 | 1.449 | 0.007 | 0.021 |
| PIK3CA | NFKBIA | 27 | 0.897 | 0.008 | 0.023 |
| AKT3 | NFKB1 | 6 | 1.922 | 0.008 | 0.023 |
| PIK3CB | AKT2 | 17 | 0.982 | 0.012 | 0.03 |
| AKT2 | MTOR | 13 | 1.12 | 0.013 | 0.031 |
| PIK3CA | IKBKB | 68 | 0.501 | 0.013 | 0.031 |
| NFKB1 | NFKBIA | 4 | 2.245 | 0.014 | 0.032 |
| RELA | NFKBIA | 6 | 1.728 | 0.014 | 0.032 |
| RELA | IKBKB | 12 | 1.106 | 0.018 | 0.04 |
| AKT1 | AKT3 | 11 | 1.129 | 0.02 | 0.043 |
| RELA | NFKB1 | 4 | 2.051 | 0.021 | 0.045 |
| RELA | NFKB2 | 4 | 1.945 | 0.026 | 0.054 |
| PIK3CA | NFKB2 | 16 | 0.96 | 0.027 | 0.054 |
| PIK3CB | PTEN | 18 | 0.772 | 0.032 | 0.062 |
| AKT3 | RELA | 8 | 1.176 | 0.036 | 0.068 |
| Drug | Target | Clinical Trial | NCT Trial |
|---|---|---|---|
| CUDC-907 | PI3K/HDAC | Phase 1 | NCT02307240 |
| Copanlisib | PI3K | Phase 1b | NCT03586661 |
| BKM120/BYL719 | PI3K | Phase 1 | NCT01623349 |
| MK2206 | AKT | Phase 2 | NCT01283035 |
| BKM120 + MEK162 | PI3K | Phase 1 | NCT01363232 |
| AZD5363 | AKT | Phase 1 | NCT01226316 |
| GSK2141795 | AKT | Phase 1 | NCT01266954 |
| Triciribine (PTX-200) | AKT | Phase 1 | NCT01690468 |
| Miransertib(ARQ 092) | AKT | Phase 1b | NCT02476955 |
| Perifosine | PI3K/ AKT | Phase 1 | NCT00431054 |
| Sapanisertib | mTOR | Phase 2 | NCT02465060 |
| Everolimus | mTOR | Phase 2 | NCT01149434 |
| Nanoparticle albumin-bound rapamycin | mTOR | Phase 1 | NCT02646319 |
| Sirolimus | mTOR | Phase 1 | NCT02833506 |
| TAK228 | PI3K/ AKT/mTOR | Phase 2 | NCT03648489 |
| MLN0128 | TORC 1/2 | Phase 1 | NCT02142803 |
| Temsirolimus | mTOR | Phase 2 | NCT02093598 |
| AZD2014 | mTOR | Phase 1b | NCT02208375 |
| IMX-110 | Stat3/NF-kB/poly-tyrosine kinase | Phase 1/2a | NCT03382340 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoneum, A.; Said, N. PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers 2019, 11, 949. https://doi.org/10.3390/cancers11070949
Ghoneum A, Said N. PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers. 2019; 11(7):949. https://doi.org/10.3390/cancers11070949
Chicago/Turabian StyleGhoneum, Alia, and Neveen Said. 2019. "PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics" Cancers 11, no. 7: 949. https://doi.org/10.3390/cancers11070949
APA StyleGhoneum, A., & Said, N. (2019). PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers, 11(7), 949. https://doi.org/10.3390/cancers11070949