The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair


Abstract
:1. Introduction
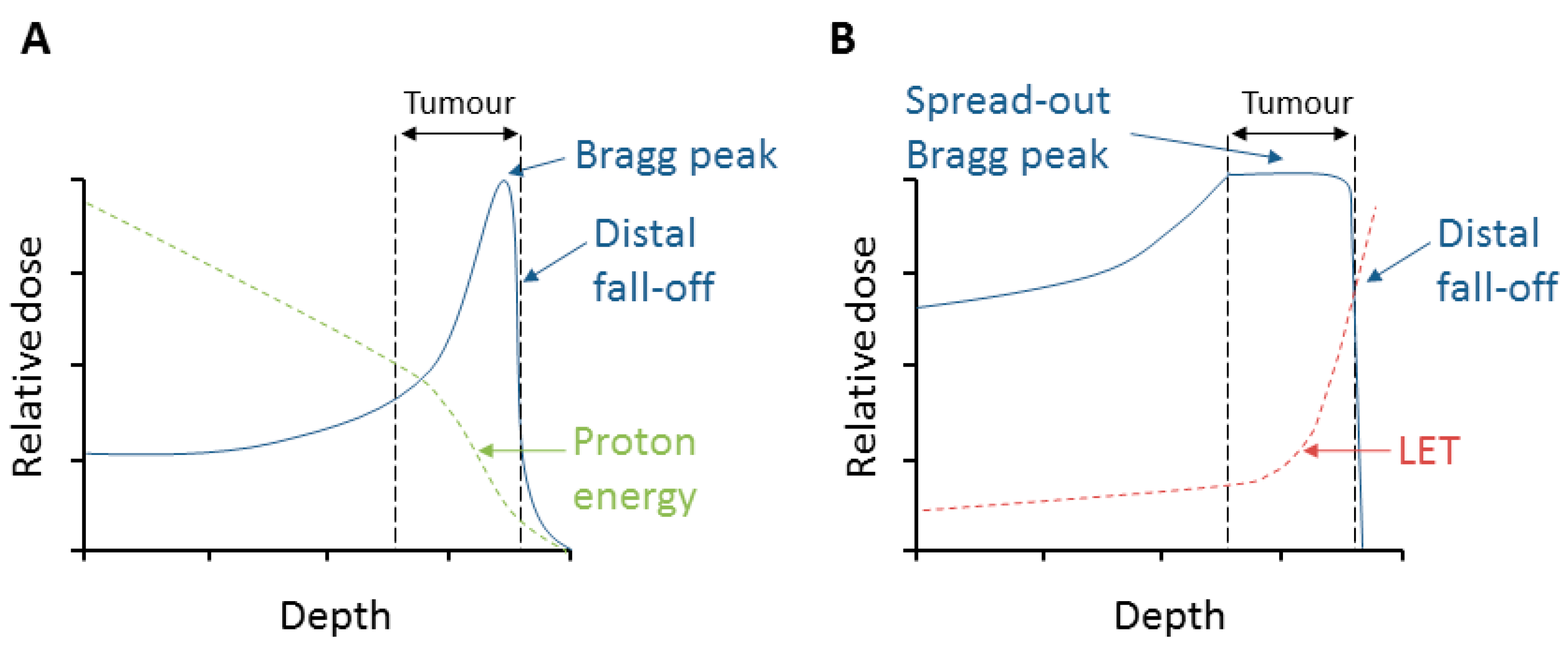
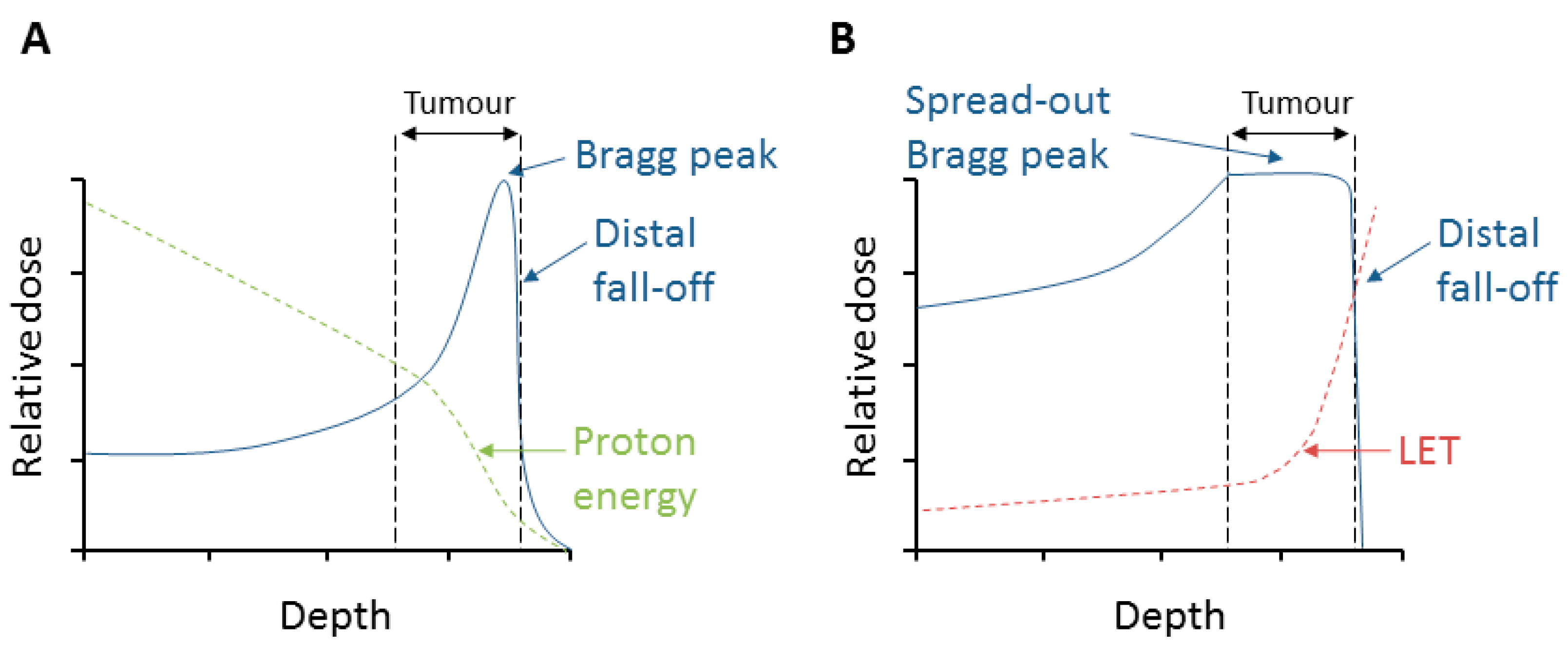
2. Relative Biological Effectiveness (RBE) and Linear Energy Transfer (LET)
3. Radiobiological Effects of Protons
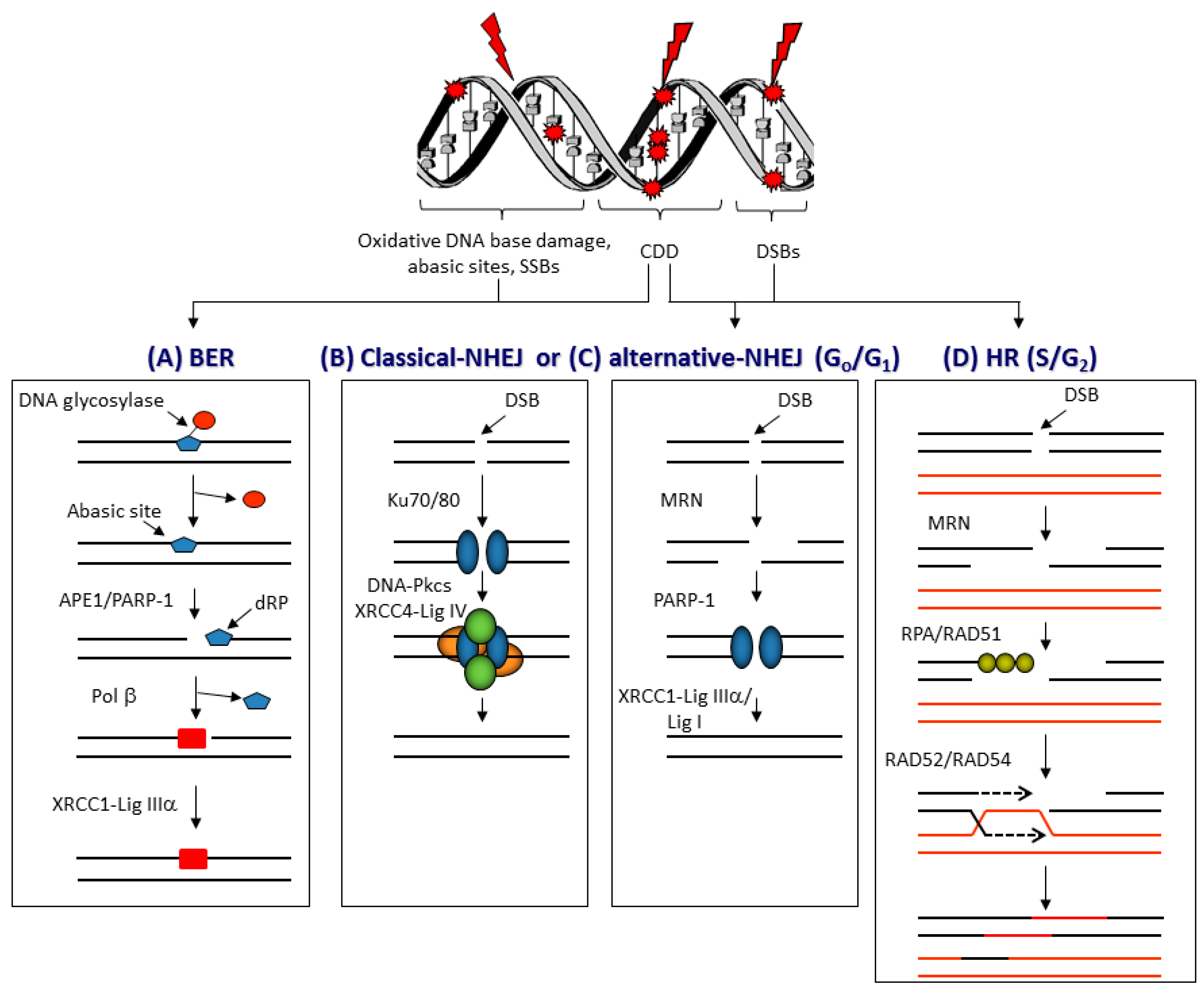
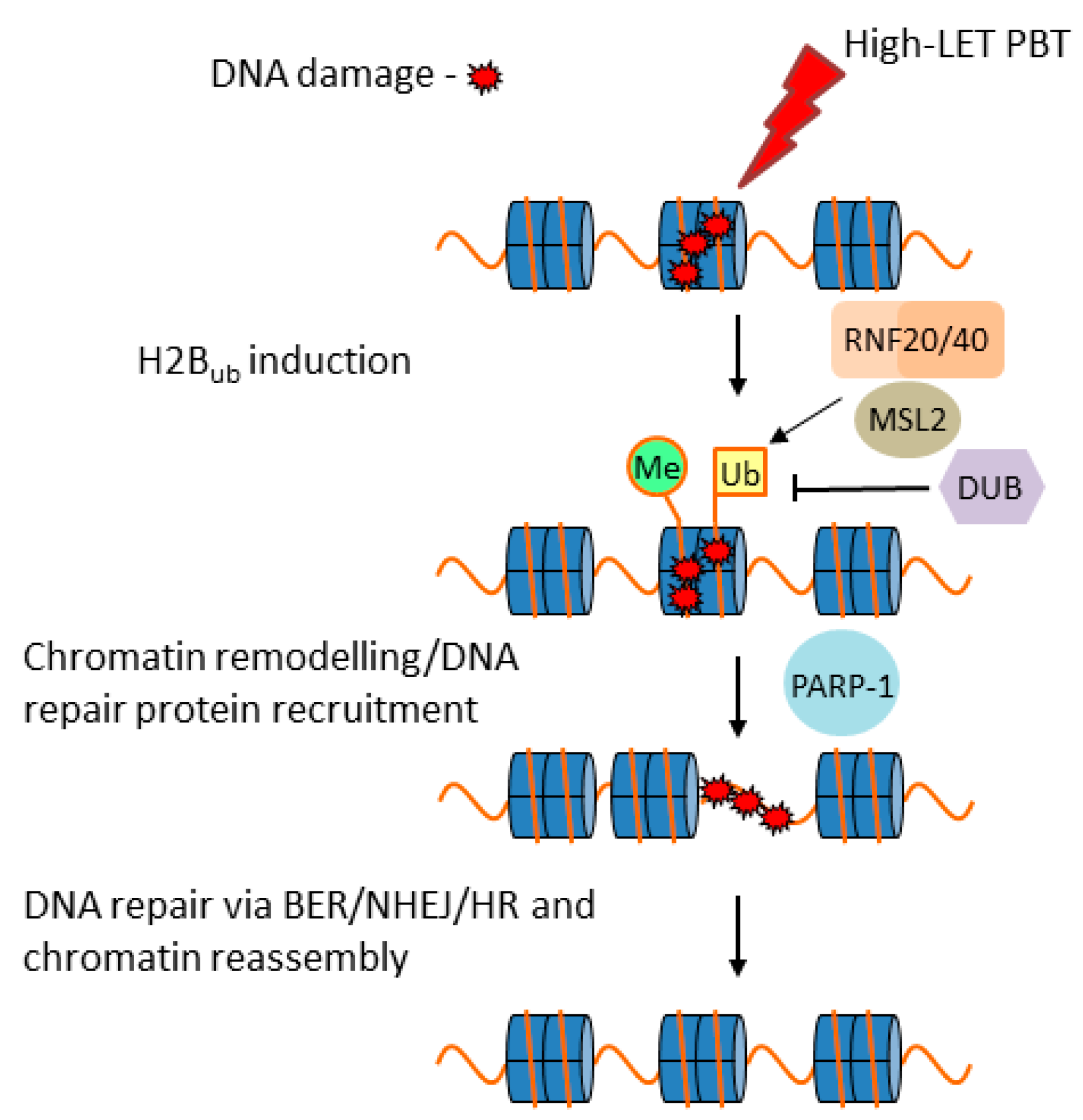
3.1. DNA Damage and Repair
3.2. DNA Damage Induction and Repair Following PBT
3.3. Generation of Reactive Oxygen Species and Cell-Cycle Progression Following PBT
3.4. DSB Repair Pathway Choice Following PBT
3.5. CDD Formation Following PBT
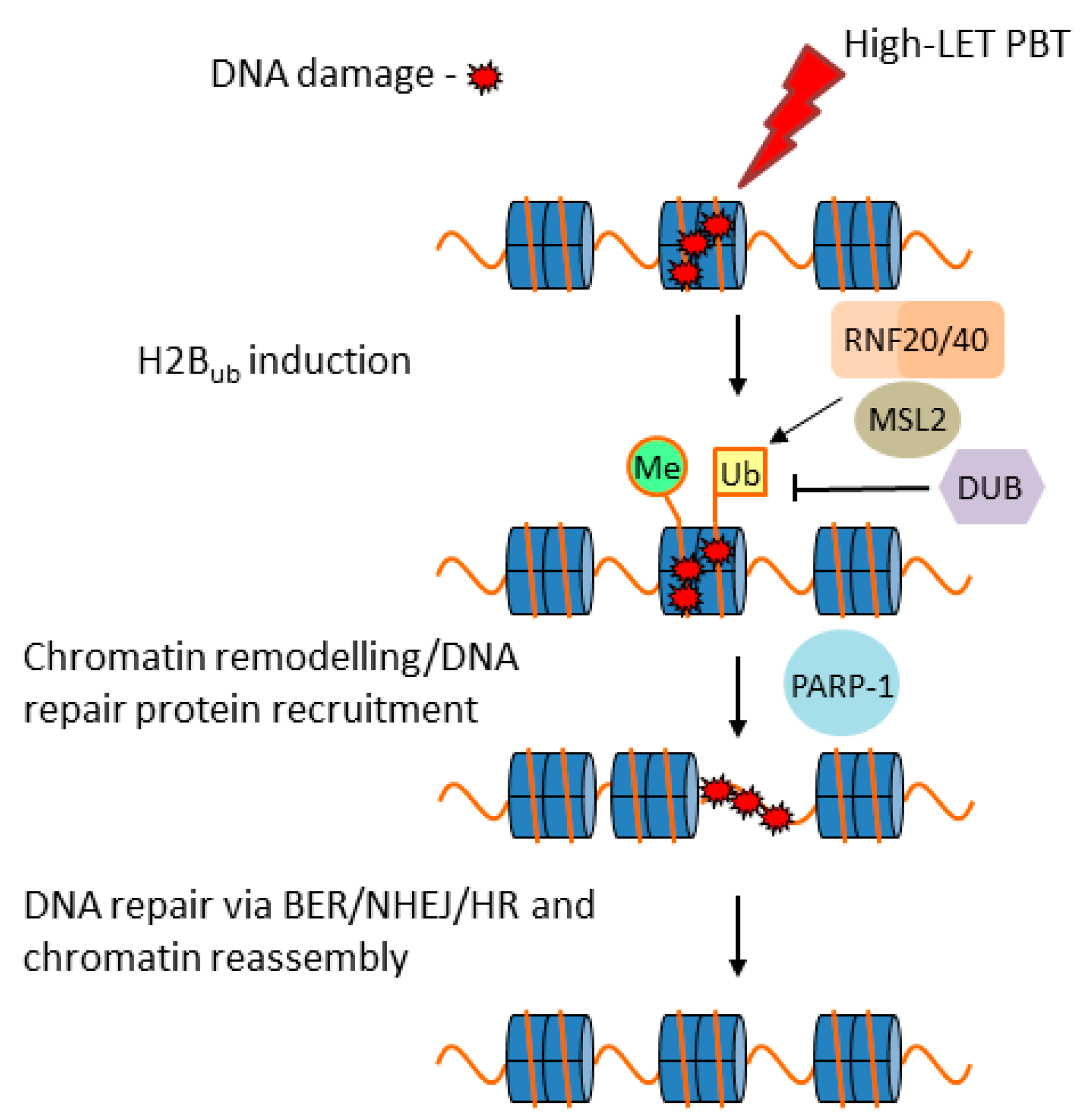
3.6. Cellular Response to CDD Generated by PBT
4. Conclusions and Outlook
- Further understanding of the biological effect of PBT at different energy/LET on the cellular DDR;
- Employing additional in vitro models (e.g., 3D spheroids/organoids) in radiobiology experiments;
- Increased utilization of in vivo experiments employing specific tumor models;
- Consideration of other biological factors (e.g., hypoxia, tumor microenvironment).
Author Contributions
Funding
Conflicts of Interest
References
- Durante, M.; Loeffler, J.S. Charged particles in radiation oncology. Nat. Rev. Clin. Oncol. 2010, 7, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, F.; Durante, M. Proton radiobiology. Cancers 2015, 7, 353–381. [Google Scholar] [CrossRef] [PubMed]
- Paganetti, H.; van Luijk, P. Biological considerations when comparing proton therapy with photon therapy. Semin. Radiat. Oncol. 2013, 23, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Cometto, A.; Russo, G.; Bourhaleb, F.; Milian, F.M.; Giordanengo, S.; Marchetto, F.; Cirio, R.; Attili, A. Direct evaluation of radiobiological parameters from clinical data in the case of ion beam therapy: An alternative approach to the relative biological effectiveness. Phys. Med. Biol. 2014, 59, 7393–7417. [Google Scholar] [CrossRef] [PubMed]
- Paganetti, H.; Niemierko, A.; Ancukiewicz, M.; Gerweck, L.E.; Goitein, M.; Loeffler, J.S.; Suit, H.D. Relative biological effectiveness (RBE) values for proton beam therapy. Int. J. Radiat. Oncol. Biol. Phys. 2002, 53, 407–421. [Google Scholar] [CrossRef]
- Carante, M.P.; Ballarini, F. Calculating variations in biological effectiveness for a 62 MeV proton beam. Front. Oncol. 2016, 6, 76. [Google Scholar] [CrossRef] [PubMed]
- Bettega, D.; Calzolari, P.; Chauvel, P.; Courdi, A.; Herault, J.; Iborra, N.; Marchesini, R.; Massariello, P.; Poli, G.L.; Tallone, L. Radiobiological studies on the 65 MeV therapeutic proton beam at Nice using human tumour cells. Int. J. Radiat. Biol. 2000, 76, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Vassiliev, O.N. Uncertainties in the relative biological effectiveness of therapeutic proton beams associated with bias towards high doses per fraction in radiobiological experiments. Int. J. Cancer Ther. Oncol. 2017, 4, 446. [Google Scholar] [CrossRef]
- Calugaru, V.; Nauraye, C.; Noel, G.; Giocanti, N.; Favaudon, V.; Megnin-Chanet, F. Radiobiological characterization of two therapeutic proton beams with different initial energy spectra used at the Institut Curie Proton Therapy Center in Orsay. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1136–1143. [Google Scholar] [CrossRef]
- Loeffler, J.S.; Durante, M. Charged particle therapy–optimization, challenges and future directions. Nat. Rev. Clin. Oncol. 2013, 10, 411–424. [Google Scholar] [CrossRef]
- Paganetti, H. Relative biological effectiveness (RBE) values for proton beam therapy. Variations as a function of biological endpoint, dose, and linear energy transfer. Phys. Med. Biol. 2014, 59, R419–R472. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, J.J.; Oelfke, U. A phenomenological model for the relative biological effectiveness in therapeutic proton beams. Phys. Med. Biol. 2004, 49, 2811–2825. [Google Scholar] [CrossRef] [PubMed]
- Britten, R.A.; Nazaryan, V.; Davis, L.K.; Klein, S.B.; Nichiporov, D.; Mendonca, M.S.; Wolanski, M.; Nie, X.; George, J.; Keppel, C. Variations in the RBE for cell killing along the depth-dose profile of a modulated proton therapy beam. Radiat. Res. 2013, 179, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Marshall, T.I.; Perozziello, F.M.; Manti, L.; Currell, F.J.; Hanton, F.; McMahon, S.J.; Kavanagh, J.N.; Cirrone, G.A.; Romano, F.; et al. Relative biological effectiveness variation along monoenergetic and modulated Bragg peaks of a 62-MeV therapeutic proton beam: A preclinical assessment. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Matsuura, T.; Wada, M.; Egashira, Y.; Nishio, T.; Furusawa, Y. Enhanced radiobiological effects at the distal end of a clinical proton beam: In vitro study. J. Radiat. Res. 2014, 55, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Saager, M.; Peschke, P.; Brons, S.; Debus, J.; Karger, C.P. Determination of the proton RBE in the rat spinal cord: Is there an increase towards the end of the spread-out Bragg peak? Radiother. Oncol. 2018, 128, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Allen, A.; Grosshans, D.; McMahon, S.J.; von Neubeck, C.; Wiese, C.; Vikram, B. Toward A variable RBE for proton beam therapy. Radiother. Oncol. 2018, 128, 68–75. [Google Scholar] [CrossRef]
- Mohan, R.; Peeler, C.R.; Guan, F.; Bronk, L.; Cao, W.; Grosshans, D.R. Radiobiological issues in proton therapy. Acta Oncol. 2017, 56, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Goodhead, D.T. Energy deposition stochastics and track structure: What about the target? Radiat. Prot. Dosim. 2006, 122, 3–15. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. 2011, 711, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. R. Coll. Radiol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef]
- Carter, R.J.; Parsons, J.L. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell. Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef] [Green Version]
- Dianov, G.L.; Parsons, J.L. Co-ordination of DNA single strand break repair. DNA Repair 2007, 6, 454–460. [Google Scholar] [CrossRef]
- Newhauser, W.D.; Zhang, R. The physics of proton therapy. Phys. Med. Biol. 2015, 60, R155–R209. [Google Scholar] [CrossRef]
- Girdhani, S.; Sachs, R.; Hlatky, L. Biological effects of proton radiation: What we know and don’t know. Radiat. Res. 2013, 179, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Hong, Z.; Sun, L.; Suzuki, K.; Terunuma, T.; Yasuoka, K.; Sakae, T.; Moritake, T.; Tsuboi, K. Induction of in situ DNA double-strand breaks and apoptosis by 200 MeV protons and 10 MV X-rays in human tumour cell lines. Int. J. Radiat. Biol. 2011, 87, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci. Rep. 2015, 5, 13961. [Google Scholar] [CrossRef] [PubMed]
- Oeck, S.; Szymonowicz, K.; Wiel, G.; Krysztofiak, A.; Lambert, J.; Koska, B.; Iliakis, G.; Timmermann, B.; Jendrossek, V. Relating linear energy transfer to the formation and resolution of DNA repair foci after irradiation with equal doses of X-ray photons, plateau, or Bragg-peak protons. Int. J. Mol. Sci. 2018, 19, 3779. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Marshall, T.I.; Currell, F.J.; Kacperek, A.; Schettino, G.; Prise, K.M. Variations in the processing of DNA double-strand breaks along 60-MeV therapeutic proton beams. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Grosse, N.; Fontana, A.O.; Hug, E.B.; Lomax, A.; Coray, A.; Augsburger, M.; Paganetti, H.; Sartori, A.A.; Pruschy, M. Deficiency in homologous recombination renders mammalian cells more sensitive to proton versus photon irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Complex DNA damage induced by high linear energy transfer alpha-particles and protons triggers a specific cellular DNA damage response. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Giedzinski, E.; Rola, R.; Fike, J.R.; Limoli, C.L. Efficient production of reactive oxygen species in neural precursor cells after exposure to 250 MeV protons. Radiat. Res. 2005, 164, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, S.H.; Fang, B.; Gillin, M.; Mohan, R.; Chang, J.Y. Therapy-resistant cancer stem cells have differing sensitivity to photon versus proton beam radiation. J. Thorac. Oncol. 2013, 8, 1484–1491. [Google Scholar] [CrossRef]
- Keta, O.D.; Todorovic, D.V.; Bulat, T.M.; Cirrone, P.G.; Romano, F.; Cuttone, G.; Petrovic, I.M.; Ristic Fira, A.M. Comparison of human lung cancer cell radiosensitivity after irradiations with therapeutic protons and carbon ions. Exp. Biol. Med. 2017, 242, 1015–1024. [Google Scholar] [CrossRef]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Characterisation of deubiquitylating enzymes in the cellular response to high-LET ionising radiation and complex DNA damage. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Moertel, H.; Georgi, J.C.; Distel, L.; Eyrich, W.; Fritsch, M.; Grabenbauer, G.; Sauer, R. Effects of low energy protons on clonogenic survival, DSB repair and cell cycle in human glioblastoma cells and B14 fibroblasts. Radiother. Oncol. 2004, 73 (Suppl. 2), S115–S118. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat. Res. 2015, 183, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Bracalente, C.; Ibanez, I.L.; Molinari, B.; Palmieri, M.; Kreiner, A.; Valda, A.; Davidson, J.; Duran, H. Induction and persistence of large gammaH2AX foci by high linear energy transfer radiation in DNA-dependent protein kinase-deficient cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.O.; Augsburger, M.A.; Grosse, N.; Guckenberger, M.; Lomax, A.J.; Sartori, A.A.; Pruschy, M.N. Differential DNA repair pathway choice in cancer cells after proton- and photon-irradiation. Radiother. Oncol. 2015, 116, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Reindl, J.; Drexler, G.A.; Girst, S.; Greubel, C.; Siebenwirth, C.; Drexler, S.E.; Dollinger, G.; Friedl, A.A. Nanoscopic exclusion between Rad51 and 53BP1 after ion irradiation in human HeLa cells. Phys. Biol. 2015, 12, 066005. [Google Scholar] [CrossRef]
- Liu, Q.; Ghosh, P.; Magpayo, N.; Testa, M.; Tang, S.; Gheorghiu, L.; Biggs, P.; Paganetti, H.; Efstathiou, J.A.; Lu, H.M.; et al. Lung cancer cell line screen links fanconi anemia/BRCA pathway defects to increased relative biological effectiveness of proton radiation. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 1081–1089. [Google Scholar] [CrossRef]
- Francis, Z.; Villagrasa, C.; Clairand, I. Simulation of DNA damage clustering after proton irradiation using an adapted DBSCAN algorithm. Comput. Methods Programs Biomed. 2011, 101, 265–270. [Google Scholar] [CrossRef]
- Nikjoo, H.; O’Neill, P.; Wilson, W.E.; Goodhead, D.T. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat. Res. 2001, 156, 577–583. [Google Scholar] [CrossRef]
- Watanabe, R.; Rahmanian, S.; Nikjoo, H. Spectrum of radiation-induced clustered non-DSB damage—A Monte Carlo track structure modeling and calculations. Radiat. Res. 2015, 183, 525–540. [Google Scholar] [CrossRef]
- Henthorn, N.T.; Warmenhoven, J.W.; Sotiropoulos, M.; Mackay, R.I.; Kirkby, N.F.; Kirkby, K.J.; Merchant, M.J. In silico non-homologous end joining following ion induced DNA double strand breaks predicts that repair fidelity depends on break density. Sci. Rep. 2018, 8, 2654. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A route to radiation-induced genomic instability and carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, C.; Sun, F.; Wei, W.; Hu, B.; Wang, J. Both complexity and location of DNA damage contribute to cellular senescence induced by ionizing radiation. PLoS ONE 2016, 11, e0155725. [Google Scholar] [CrossRef] [PubMed]
- Asaithamby, A.; Hu, B.; Chen, D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8293–8298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2016, 107, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Zlobinskaya, O.; Siebenwirth, C.; Greubel, C.; Hable, V.; Hertenberger, R.; Humble, N.; Reinhardt, S.; Michalski, D.; Roper, B.; Multhoff, G.; et al. The effects of ultra-high dose rate proton irradiation on growth delay in the treatment of human tumor xenografts in nude mice. Radiat. Res. 2014, 181, 177–183. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Cell Line | Method(s) | Proton Energy | Photon Energy | Observation (Proton vs. Photon) | Ref |
|---|---|---|---|---|---|
| ONS76 medulloblastoma; MOLT4 leukemia cells | γH2AX foci by immunofluorescence | 200 MeV | 10 MV X-rays | ~1.2–1.6-fold increase in DSB foci and ~1.2–1.5-fold larger in size 30–180 min post-irradiation | [32] |
| HeLa; SQ20B HNSCC cells | Pulse-field gel electrophoresis | 76 MeV, 201 MeV | 622 keV 137Cs γ-rays | ~1.2-fold increase in DSBs. No differences between PBT energies, nor along the SOBP | [9] |
| IN528 and T4213 glioblastoma stem-like cells | Alkaline and neutral comet assay | N.S. | 320 kV X-rays | ~1.2–1.6-fold higher numbers of DSBs at 20–48 h post-irradiation | [33] |
| TrC1 prostate cancer cells; murine embryonic fibroblasts | Histone γH2AX and 53BP1 foci by immunofluorescence | 187 MeV entrance dose | 320 kV X-rays | Similar numbers of DSBs at 0.5–24 h post-irradiation | [34] |
| AG01522 skin fibroblasts | 53BP1 foci by immunofluorescence | 60 MeV entrance dose | 225 kV X-rays | Similar numbers of DSBs at 0.5–24 h post-irradiation | [35] |
| Wild-type, HR-, and NHEJ-deficient Chinese hamster ovary cell lines | Histone γH2AX foci by immunofluorescence | 138 MeV | 200 kV X-rays | Similar initial induction of DSBs | [36] |
| HeLa; UMSCC74A and UMSCC6 HNSCC cells | Neutral comet assay | 58 MeV entrance dose; 11 MeV distal edge | 100 kV X-rays | No difference in DSB repair kinetics | [37] |
| Cell line | Irradiations | Outcome | Ref |
|---|---|---|---|
| Wild-type, HR- and NHEJ-deficient Chinese hamster ovary cell lines | 200 MeV protons and 137Cs γ-rays | NHEJ is the major pathway for both photons and low-LET protons | [43] |
| Wild-type and NHEJ-deficient Chinese hamster ovary cell lines | 14.4 MeV plateau protons and 667 keV 137Cs γ-rays | NHEJ is the major pathway for both photons and low-LET protons | [44] |
| Wild-type, HR-, and NHEJ-deficient Chinese hamster ovary cell lines | 138 MeV protons and 200-kV X-rays | Dependence on HR following protons | [36] |
| A549 lung cancer; glioblastoma cells | 138 MeV protons and 200 kV X-rays | Dependence on HR following protons | [45] |
| HeLa | 21 MeV protons | Higher proportion of cells undergoing HR following protons | [46] |
| Non-small-cell lung cancer cells | 235 MeV protons and 250 kV X-rays | HR only partly required following protons | [47] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitti, E.T.; Parsons, J.L. The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair. Cancers 2019, 11, 946. https://doi.org/10.3390/cancers11070946
Vitti ET, Parsons JL. The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair. Cancers. 2019; 11(7):946. https://doi.org/10.3390/cancers11070946
Chicago/Turabian StyleVitti, Eirini Terpsi, and Jason L Parsons. 2019. "The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair" Cancers 11, no. 7: 946. https://doi.org/10.3390/cancers11070946
APA StyleVitti, E. T., & Parsons, J. L. (2019). The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair. Cancers, 11(7), 946. https://doi.org/10.3390/cancers11070946