Wnt Signaling in Cancer Metabolism and Immunity


Abstract
1. Introduction
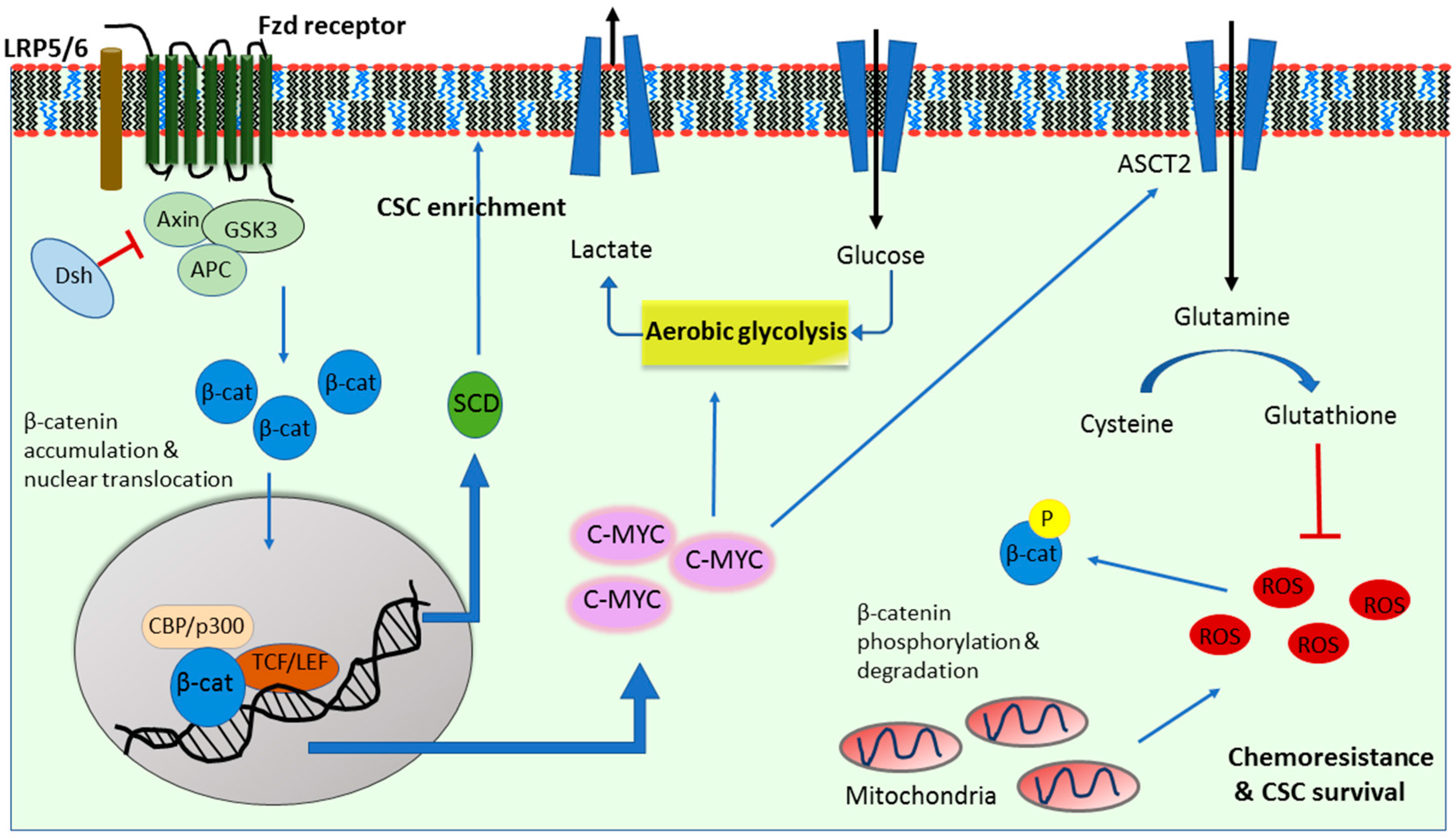
2. Wnt Signaling in Cancer Glycolysis
3. Wnt Signaling in Cancer Glutaminolysis
4. Wnt Signaling in Cancer Lipogenesis
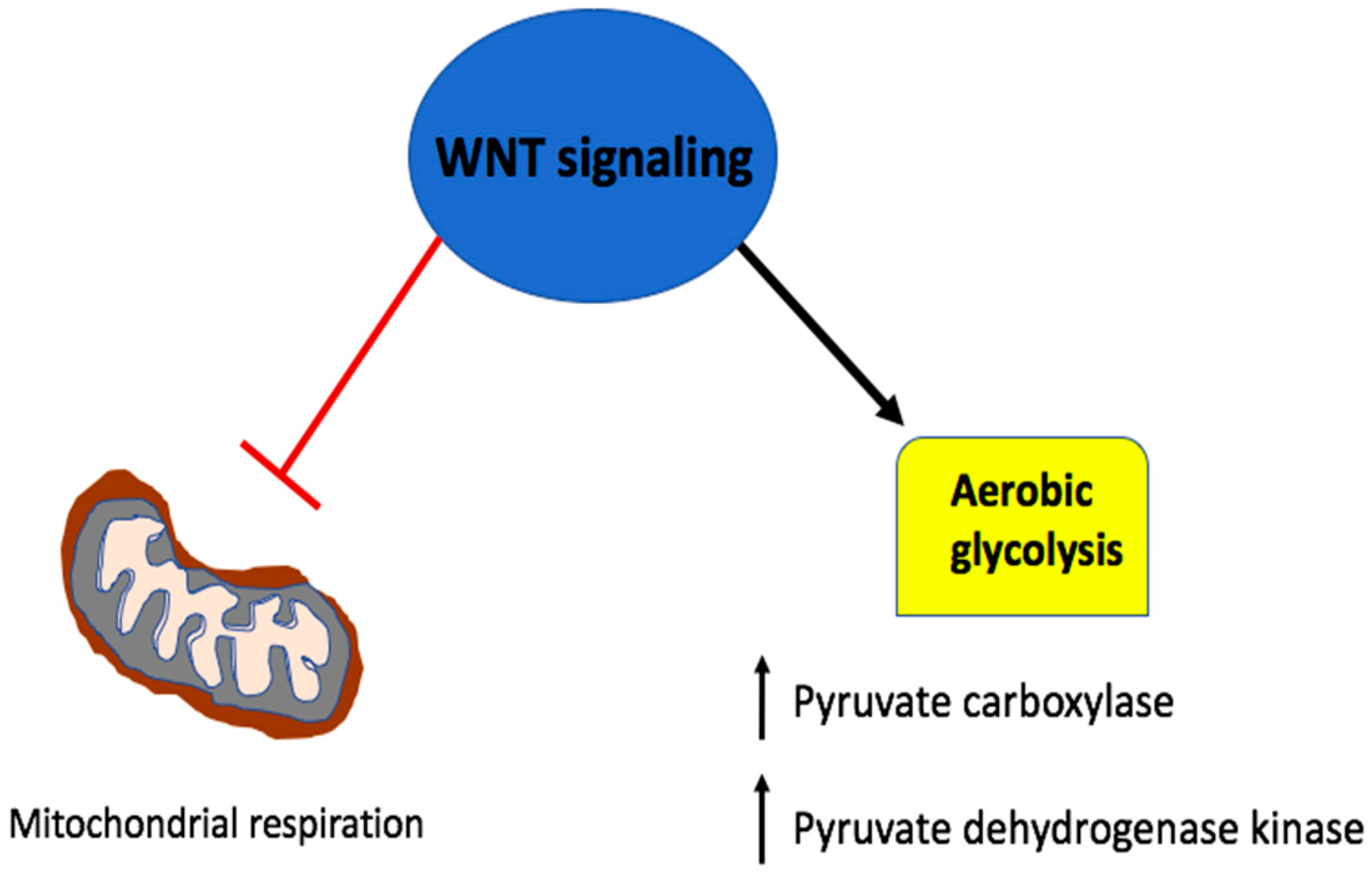
5. Wnt Signaling in A Metabolic Negative Feedback Loop
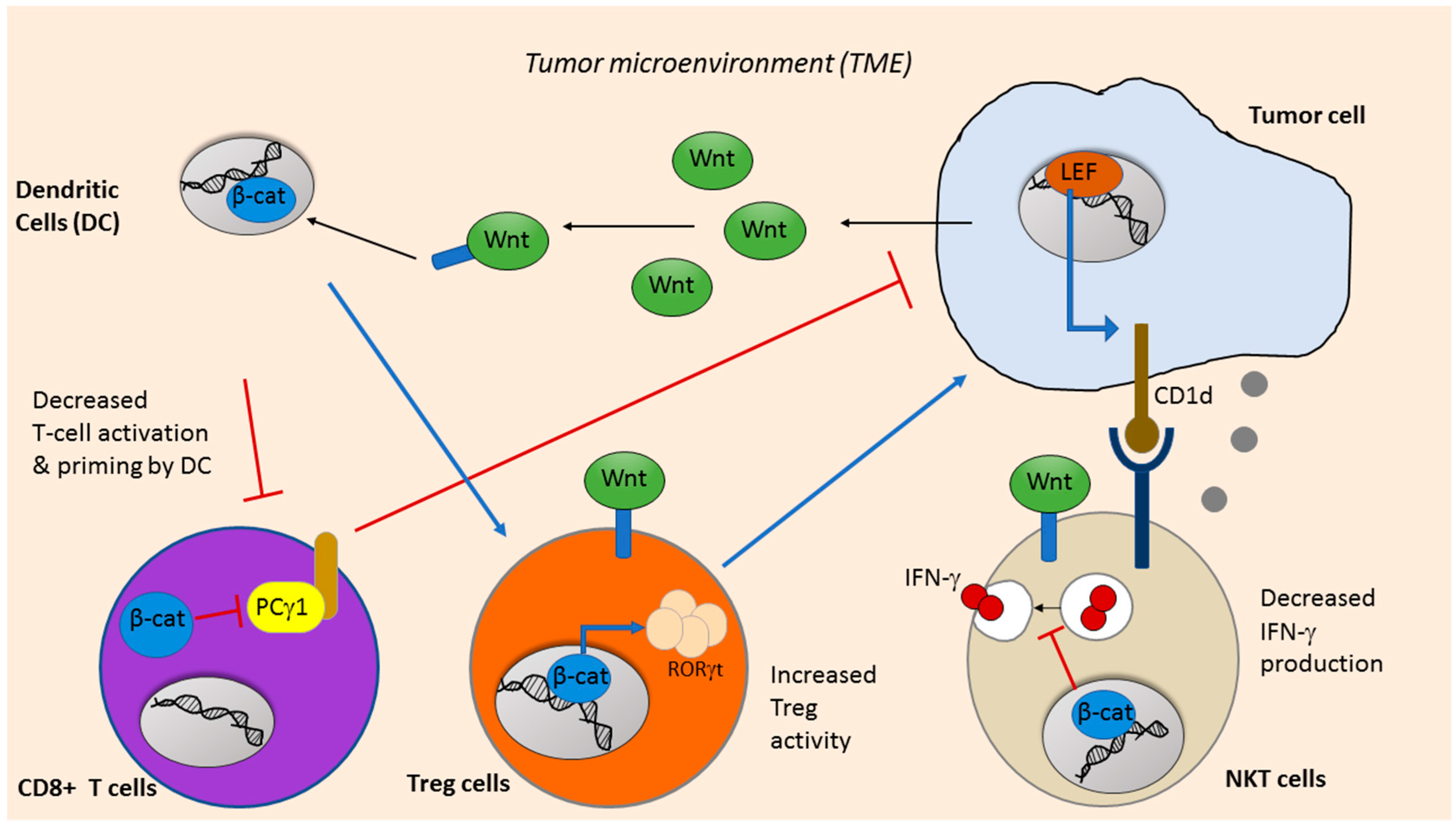
6. Wnt Signaling in Cancer Immunotherapy
7. Future Directions: Targeting Wnt Signaling to Inhibit Cancer Metabolism and Enhance Immune Response
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Tian, A.; Benchabane, H.; Wang, Z.; Ahmed, Y. Regulation of Stem Cell Proliferation and Cell Fate Specification by Wingless/Wnt Signaling Gradients Enriched at Adult Intestinal Compartment Boundaries. PLoS Genet. 2016, 12, e1005822. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Schlange, T.; Oakeley, E.J.; Boulay, A.; Hynes, N.E. WNT signaling enhances breast cancer cell motility and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231 xenograft growth. Breast Cancer Res. 2009, 11, R32. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Yang, W.; Li, L.; Liu, H.; Tan, Y.; Ooi, S.; Chi, L.; Filion, L.G.; Figeys, D.; Wang, L. Beta-Catenin and NF-kappaB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ. 2015, 22, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymaki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, P.H.; Nephew, K.P. Multiple signaling pathways converge on beta-catenin in thyroid cancer. Thyroid 2005, 15, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sinha, T.; Wynshaw-Boris, A. Wnt signaling in mammalian development: Lessons from mouse genetics. Cold Spring Harb. Perspect. Biol. 2012, 4, a007963. [Google Scholar] [CrossRef]
- Ramos-Solano, M.; Meza-Canales, I.D.; Torres-Reyes, L.A.; Alvarez-Zavala, M.; Alvarado-Ruiz, L.; Rincon-Orozco, B.; Garcia-Chagollan, M.; Ochoa-Hernandez, A.B.; Ortiz-Lazareno, P.C.; Rosl, F.; et al. Expression of WNT genes in cervical cancer-derived cells: Implication of WNT7A in cell proliferation and migration. Exp. Cell Res. 2015, 335, 39–50. [Google Scholar] [CrossRef]
- Cui, N.; Yang, W.T.; Zheng, P.S. Slug inhibits the proliferation and tumor formation of human cervical cancer cells by up-regulating the p21/p27 proteins and down-regulating the activity of the Wnt/beta-catenin signaling pathway via the trans-suppression Akt1/p-Akt1 expression. Oncotarget 2016, 7, 26152–26167. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Mohseny, A.B.; Karperien, M.; Hogendoorn, P.C.; Zhou, G.; Cleton-Jansen, A.M. Inactive Wnt/beta-catenin pathway in conventional high-grade osteosarcoma. J. Pathol. 2009, 220, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Knowles, E.; Zardawi, S.J.; McNeil, C.M.; Millar, E.K.; Crea, P.; Musgrove, E.A.; Sutherland, R.L.; O’Toole, S.A. Cytoplasmic localization of beta-catenin is a marker of poor outcome in breast cancer patients. Cancer Epidemiol. Prev. Biomark. 2010, 19, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Haussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hu, J.; Huang, Z.; Li, H.; Liu, X.; Feng, W. Effect of indomethacin on BCR/ABL-Wnt/beta-catenin pathway in K562 cells. Nan Fang Yi Ke Da Xue Xue Bao 2015, 35, 998–1002. [Google Scholar] [PubMed]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.C.; Londono-Joshi, A.I.; Gangrade, A.; Katre, A.A.; Kurpad, C.; Li, Y.; Samant, R.S.; Li, P.K.; Landen, C.N.; Yang, E.S.; et al. Niclosamide and its analogs are potent inhibitors of Wnt/beta-catenin, mTOR and STAT3 signaling in ovarian cancer. Oncotarget 2016, 7, 86803–86815. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lan, C.; Li, L.; Yang, D.; Xia, X.; Liao, Q.; Fu, W.; Chen, X.; An, S.; Wang, W.E.; et al. A novel porcupine inhibitor blocks WNT pathways and attenuates cardiac hypertrophy. Biochim Biophys. Acta Mol. Basis Dis. 2018, 1864, 3459–3467. [Google Scholar] [CrossRef]
- Huang, R.; Han, J.; Liang, X.; Sun, S.; Jiang, Y.; Xia, B.; Niu, M.; Li, D.; Zhang, J.; Wang, S.; et al. Androgen Receptor Expression and Bicalutamide Antagonize Androgen Receptor Inhibit beta-Catenin Transcription Complex in Estrogen Receptor-Negative Breast Cancer. Cell Physiol. Biochem. 2017, 43, 2212–2225. [Google Scholar] [CrossRef]
- Mina, A.; Yoder, R.; Sharma, P. Targeting the androgen receptor in triple-negative breast cancer: Current perspectives. Onco Targets Ther. 2017, 10, 4675–4685. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Schoffski, P.; Littlefield, B.A. Multiple modes of action of eribulin mesylate: Emerging data and clinical implications. Cancer Treat. Rev. 2018, 70, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Lin, X.; Zhang, C.; An, G.; Li, Z.; Dong, B.; Shen, L.; Gao, J.; Zhang, X. Activated Wnt signaling promotes growth and progression of AFP-producing gastric cancer in preclinical models. Cancer Manag. Res. 2019, 11, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, Y.; Liu, H.; Yang, J.; Song, X.; Zhao, J.; He, N.; Zhou, C.J.; Wang, Y.; Dong, Q. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget 2017, 8, 115254–115269. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Ong, R.; Goh, K.Y.; Lee, L.Y.; Puehler, F.; Scholz, A.; Politz, O.; Mumberg, D.; Ziegelbauer, K. Sorafenib/MEK inhibitor combination inhibits tumor growth and the Wnt/betacatenin pathway in xenograft models of hepatocellular carcinoma. Int. J. Oncol. 2019, 54, 1123–1133. [Google Scholar]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Cao, L.; Zhu, J.; Tan, Z.; Tang, M.; Li, Z.; Hu, Y.; Yu, R.; Zhang, S.; Song, L.; et al. Loss of RBMS3 Confers Platinum Resistance in Epithelial Ovarian Cancer via Activation of miR-126-5p/beta-catenin/CBP signaling. Clin. Cancer Res. 2019, 25, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, U.H.; Senapedis, W.; Baloglu, E.; Unger, T.J.; Chari, A.; Vogl, D.; Cornell, R.F. Clinical Implications of Targeting XPO1-mediated Nuclear Export in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 335–345. [Google Scholar] [CrossRef]
- Lu, D.; Liu, J.X.; Endo, T.; Zhou, H.; Yao, S.; Willert, K.; Schmidt-Wolf, I.G.; Kipps, T.J.; Carson, D.A. Ethacrynic acid exhibits selective toxicity to chronic lymphocytic leukemia cells by inhibition of the Wnt/beta-catenin pathway. PLoS ONE 2009, 4, e8294. [Google Scholar] [CrossRef]
- Liu, H.; Fergusson, M.M.; Wu, J.J.; Rovira, I.I.; Liu, J.; Gavrilova, O.; Lu, T.; Bao, J.; Han, D.; Sack, M.N.; et al. Wnt signaling regulates hepatic metabolism. Sci. Signal. 2011, 4, ra6. [Google Scholar] [CrossRef] [PubMed]
- Fevr, T.; Robine, S.; Louvard, D.; Huelsken, J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell Biol. 2007, 27, 7551–7559. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, M.; Reichert, T.E.; Benz, P.; Lehr, H.A.; Jeong, J.H.; Wieand, S.; Bartenstein, P.; Wagner, W.; Whiteside, T.L. Overexpression of Glut-1 and increased glucose metabolism in tumors are associated with a poor prognosis in patients with oral squamous cell carcinoma. Cancer 2003, 97, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Perez, A.A.; Fujie, S.; Warden, C.; Li, J.; Wang, Y.; Yung, B.; Chen, Y.R.; Liu, X.; Zhang, H.; et al. Wnt modulates MCL1 to control cell survival in triple negative breast cancer. BMC Cancer 2014, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4). Cancers (Basel) 2018, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lu, P.; Zhang, H.; Xu, H.; Gao, N.; Li, M.; Liu, C. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014, 16, 408. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.N.; Marks, J.R.; Chi, J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Li, C.; Zhang, G.; Zhao, L.; Ma, Z.; Chen, H. Metabolic reprogramming in cancer cells: Glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J. Surg. Oncol. 2016, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, Z.; Yang, P.; Zhang, L.; Fan, Y. PKM2 depletion induces the compensation of glutaminolysis through beta-catenin/c-Myc pathway in tumor cells. Cell. Signal. 2014, 26, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through beta-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef] [PubMed]
- Mihm, S.; Galter, D.; Droge, W. Modulation of transcription factor NF kappa B activity by intracellular glutathione levels and by variations of the extracellular cysteine supply. FASEB J. 1995, 9, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; Tannir, N.M.; Owonikoko, T.K.; Haas, N.B.; Voss, M.H.; et al. CB-839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase I study. J. Clin. Oncol. 2019, 37, 549. [Google Scholar] [CrossRef]
- Jones, J.E.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo-Calvo, D.; Lopez-Vilaro, L.; Nasarre, L.; Perez-Olabarria, M.; Vazquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortes, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Hardy, R.W.; Ren, C.; Ponnazhagan, S.; Welch, D.R.; McDonald, J.M.; Chen, Y. Free fatty acids enhance breast cancer cell migration through plasminogen activator inhibitor-1 and SMAD4. Lab. Investig. 2009, 89, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Sun, S.; Wang, J.; Fei, F.; Dong, Z.; Ke, A.W.; He, R.; Wang, L.; Zhang, L.; Ji, M.B.; et al. Canonical Wnt Signaling Remodels Lipid Metabolism in Zebrafish Hepatocytes following Ras Oncogenic Insult. Cancer Res. 2018, 78, 5548–5560. [Google Scholar] [CrossRef] [PubMed]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.P.; Fargnoli, J.; Henley, B.J.; Yang, W.P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Kenny, H.A.; Lengyel, E. Unsaturated Fatty Acids Maintain Cancer Cell Stemness. Cell Stem Cell 2017, 20, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Stanca, E.; Guerra, F.; Priore, P.; Gaballo, A.; Franck, J.; Simeone, P.; Trerotola, M.; De Domenico, S.; Fournier, I.; et al. beta-Catenin Knockdown Affects Mitochondrial Biogenesis and Lipid Metabolism in Breast Cancer Cells. Front. Physiol. 2017, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150.e5. [Google Scholar] [CrossRef]
- Colacino, J.A.; McDermott, S.P.; Sartor, M.A.; Wicha, M.S.; Rozek, L.S. Transcriptomic profiling of curcumin-treated human breast stem cells identifies a role for stearoyl-coa desaturase in breast cancer prevention. Breast Cancer Res. Treat. 2016, 158, 29–41. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Lai, K.K.Y.; Kweon, S.M.; Chi, F.; Hwang, E.; Kabe, Y.; Higashiyama, R.; Qin, L.; Yan, R.; Wu, R.P.; Lai, K.; et al. Stearoyl-CoA Desaturase Promotes Liver Fibrosis and Tumor Development in Mice via a Wnt Positive-Signaling Loop by Stabilization of Low-Density Lipoprotein-Receptor-Related Proteins 5 and 6. Gastroenterology 2017, 152, 1477–1491. [Google Scholar] [CrossRef] [PubMed]
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013, 4, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; McGarry, S.; Li, L.; Jia, D.; Ooi, S.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; Nessim, C.; Yao, Z. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, L.; Zhang, H.; Wang, D.; Wang, M.; Zhu, J.; Pang, C.; Wang, C. Succinate dehydrogenase 5 (SDH5) regulates glycogen synthase kinase 3beta-beta-catenin-mediated lung cancer metastasis. J. Biol. Chem. 2013, 288, 29965–29973. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Takemaru, K.; Yamaguchi, S.; Lee, Y.S.; Zhang, Y.; Carthew, R.W.; Moon, R.T. Chibby, a nuclear beta-catenin-associated antagonist of the Wnt/Wingless pathway. Nature 2003, 422, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.F.; Ye, G.D.; Shen, D.Y.; Zhang, W.; Chen, M.L.; Chen, X.X.; Han, D.X.; Mi, Y.J.; Luo, Q.C.; Cai, W.Y.; et al. Chibby suppresses aerobic glycolysis and proliferation of nasopharyngeal carcinoma via the Wnt/beta-catenin-Lin28/let7-PDK1 cascade. J. Exp. Clin. Cancer Res. 2018, 37, 104. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.A.; Xiong, X.; Scott, T.; Li, A.T.; Wang, C.; Weiss, H.L.; Tan, L.; Bradford, E.; Fan, T.W.M.; Chandel, N.S.; et al. The mitochondrial retrograde signaling regulates Wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Shi, Y.B. An essential and evolutionarily conserved role of protein arginine methyltransferase 1 for adult intestinal stem cells during postembryonic development. Stem Cells 2010, 28, 2073–2083. [Google Scholar] [CrossRef]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2014, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed]
- den Haan, J.M.; Lehar, S.M.; Bevan, M.J. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Hu, J. Role of dendritic cells in the induction of regulatory T cells. Cell Biosci. 2011, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Fu, C.; Cui, W.; Ober-Blobaum, J.L.; Zahner, S.P.; Shrikant, P.A.; Clausen, B.E.; Flavell, R.A.; Mellman, I.; Jiang, A. beta-catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8(+) T cells. J. Leukoc. Biol. 2014, 95, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.C.; Pulendran, B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Shanmugam, A.; Swafford, D.; Ahmad, S.; Chinnadurai, R.; Manicassamy, B.; He, Y.; Mellor, A.L.; et al. Deletion of LRP5 and LRP6 in dendritic cells enhances antitumor immunity. Oncoimmunology 2016, 5, e1115941. [Google Scholar] [CrossRef]
- Kerdidani, D.; Chouvardas, P.; Arjo, A.R.; Giopanou, I.; Ntaliarda, G.; Guo, Y.A.; Tsikitis, M.; Kazamias, G.; Potaris, K.; Stathopoulos, G.T.; et al. Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma. Nat. Commun. 2019, 10, 1405. [Google Scholar] [CrossRef] [PubMed]
- van Loosdregt, J.; Fleskens, V.; Tiemessen, M.M.; Mokry, M.; van Boxtel, R.; Meerding, J.; Pals, C.E.; Kurek, D.; Baert, M.R.; Delemarre, E.M.; et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 2013, 39, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Keerthivasan, S.; Aghajani, K.; Dose, M.; Molinero, L.; Khan, M.W.; Venkateswaran, V.; Weber, C.; Emmanuel, A.O.; Sun, T.; Bentrem, D.J.; et al. beta-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci. Transl. Med. 2014, 6, 225ra28. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Jin, J.Q.; Xia, L.; Xiao, T.; Mei, S.; Wang, X.; Huang, X.; Chen, J.; Liu, M.; Chen, C.; et al. Pharmacological inhibition of beta-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating Treg cells. Sci. Adv. 2019, 5, eaau5240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.M.; Yu, S.; Zhou, X.; Haring, J.S.; Held, W.; Badovinac, V.P.; Harty, J.T.; Xue, H.H. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J. Immunol. 2010, 184, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Driessens, G.; Zheng, Y.; Locke, F.; Cannon, J.L.; Gounari, F.; Gajewski, T.F. Beta-catenin inhibits T cell activation by selective interference with linker for activation of T cells-phospholipase C-gamma1 phosphorylation. J. Immunol. 2011, 186, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Wu, J.; Wang, W.J.; Chen, S.; Zheng, Y.; Yu, X.; Meeth, K.; Sahraei, M.; Bothwell, A.L.M.; Chen, L.; et al. DKK2 imparts tumor immunity evasion through beta-catenin-independent suppression of cytotoxic immune-cell activation. Nat. Med. 2018, 24, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Dhodapkar, M.V. Natural Killer T Cells in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Zhang, T.; Pincus, S.H.; Wu, S.; Ricks, D.; Liu, D.; Sun, Z.; Maclaren, N.; Lan, M.S. Human CD1D gene expression is regulated by LEF-1 through distal promoter regulatory elements. J. Immunol. 2010, 184, 5047–5054. [Google Scholar] [CrossRef] [PubMed]
- Berga-Bolanos, R.; Sharma, A.; Steinke, F.C.; Pyaram, K.; Kim, Y.H.; Sultana, D.A.; Fang, J.X.; Chang, C.H.; Xue, H.H.; Heller, N.M.; et al. beta-Catenin is required for the differentiation of iNKT2 and iNKT17 cells that augment IL-25-dependent lung inflammation. BMC Immunol. 2015, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Pyaram, K.; Sen, J.M.; Chang, C.H. Temporal regulation of Wnt/beta-catenin signaling is important for invariant NKT cell development and terminal maturation. Mol. Immunol. 2017, 85, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kling, J.C.; Jordan, M.A.; Pitt, L.A.; Meiners, J.; Thanh-Tran, T.; Tran, L.S.; Nguyen, T.T.K.; Mittal, D.; Villani, R.; Steptoe, R.J.; et al. Temporal Regulation of Natural Killer T Cell Interferon Gamma Responses by beta-Catenin-Dependent and -Independent Wnt Signaling. Front. Immunol. 2018, 9, 483. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Yu, H.; Zhang, J.; Jing, H.; Zhu, W.; Li, X.; Kong, L.; Xing, L.; Yu, J.; Meng, X. Correlation of cancer stem cell markers and immune cell markers in resected non-small cell lung cancer. J. Cancer 2017, 8, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Robitaille, A.M.; Berndt, J.D.; Davidson, K.C.; Fischer, K.A.; Mathieu, J.; Potter, J.C.; Ruohola-Baker, H.; Moon, R.T. Wnt/beta-catenin signaling promotes self-renewal and inhibits the primed state transition in naive human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6382–E6390. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Zeng, H.; Horng, T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019, 21, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2009, 70, 68–77. [Google Scholar] [CrossRef]
- Van de Velde, L.A.; Subramanian, C.; Smith, A.M.; Barron, L.; Qualls, J.E.; Neale, G.; Alfonso-Pecchio, A.; Jackowski, S.; Rock, C.O.; Wynn, T.A.; et al. T Cells Encountering Myeloid Cells Programmed for Amino Acid-dependent Immunosuppression Use Rictor/mTORC2 Protein for Proliferative Checkpoint Decisions. J. Biol. Chem. 2017, 292, 15–30. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Bellone, M.; Calcinotto, A.; Filipazzi, P.; De Milito, A.; Fais, S.; Rivoltini, L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology 2013, 2, e22058. [Google Scholar] [CrossRef] [PubMed]
- Sprowl-Tanio, S.; Habowski, A.N.; Pate, K.T.; McQuade, M.M.; Wang, K.; Edwards, R.A.; Grun, F.; Lyou, Y.; Waterman, M.L. Lactate/pyruvate transporter MCT-1 is a direct Wnt target that confers sensitivity to 3-bromopyruvate in colon cancer. Cancer Metab. 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Vincent, E.E.; Cronin, J.G.; Panetti, S.; Chambers, M.; Holm, S.R.; Owens, S.E.; Francis, N.J.; Finlay, D.K.; Thornton, C.A. Akt and STAT5 mediate naive human CD4+ T-cell early metabolic response to TCR stimulation. Nat. Commun. 2019, 10, 2042. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 2008, 283, 21934–21944. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- van der Mijn, J.C.; Panka, D.J.; Geissler, A.K.; Verheul, H.M.; Mier, J.W. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. 2016, 4, 14. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef]
- Newsholme, P.; Curi, R.; Pithon Curi, T.C.; Murphy, C.J.; Garcia, C.; Pires de Melo, M. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: Its importance in health and disease. J. Nutr. Biochem. 1999, 10, 316–324. [Google Scholar] [CrossRef]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef]
- Wang, W.; Kryczek, I.; Dostal, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Cancer Type | Phase | Component Targeted | References |
|---|---|---|---|---|
| NCT02950259 | Breast cancer | I | β-catenin | [16] |
| NCT02807805 | Prostate cancer | II | Dvl & β-catenin | [17,18] |
| NCT02675946 | GI cancer | I | Wnt ligands | [19] |
| NCT03090165 | TNBC | II | β-catenin | [20,21] |
| NCT02513472 | Breast cancer | I | β-catenin | [22] |
| NCT03355066 | Advanced solid tumors | I | Unknown | [23] |
| NCT01351103 | Lung cancer, colorectal cancer, TNBC... etc. | I | Wnt ligands | [24] |
| NCT02429427 | Breast cancer | III | GSK3 | [25] |
| NCT02346032 | Biliary Tract Cancer | II | Wnt3 & LRP6 | [26] |
| NCT02005315 | Pancreatic Cancer | I | FZD | [27] |
| NCT01302405 | Advanced solid tumors | I | β-catenin/CBP | [28] |
| NCT02402764 | Breast cancer | II | APC | [29] |
| NCT02426723 | Multiple myeloma | I | β-catenin | NCT02426723 |
| NCT02852564 | Bladder cancer | I | LEF-1/βcatenin | [30] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. https://doi.org/10.3390/cancers11070904
El-Sahli S, Xie Y, Wang L, Liu S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers. 2019; 11(7):904. https://doi.org/10.3390/cancers11070904
Chicago/Turabian StyleEl-Sahli, Sara, Ying Xie, Lisheng Wang, and Sheng Liu. 2019. "Wnt Signaling in Cancer Metabolism and Immunity" Cancers 11, no. 7: 904. https://doi.org/10.3390/cancers11070904
APA StyleEl-Sahli, S., Xie, Y., Wang, L., & Liu, S. (2019). Wnt Signaling in Cancer Metabolism and Immunity. Cancers, 11(7), 904. https://doi.org/10.3390/cancers11070904