Aberrant MicroRNA Expression and Its Implications for Uveal Melanoma Metastasis

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Sample Collection and Analysis
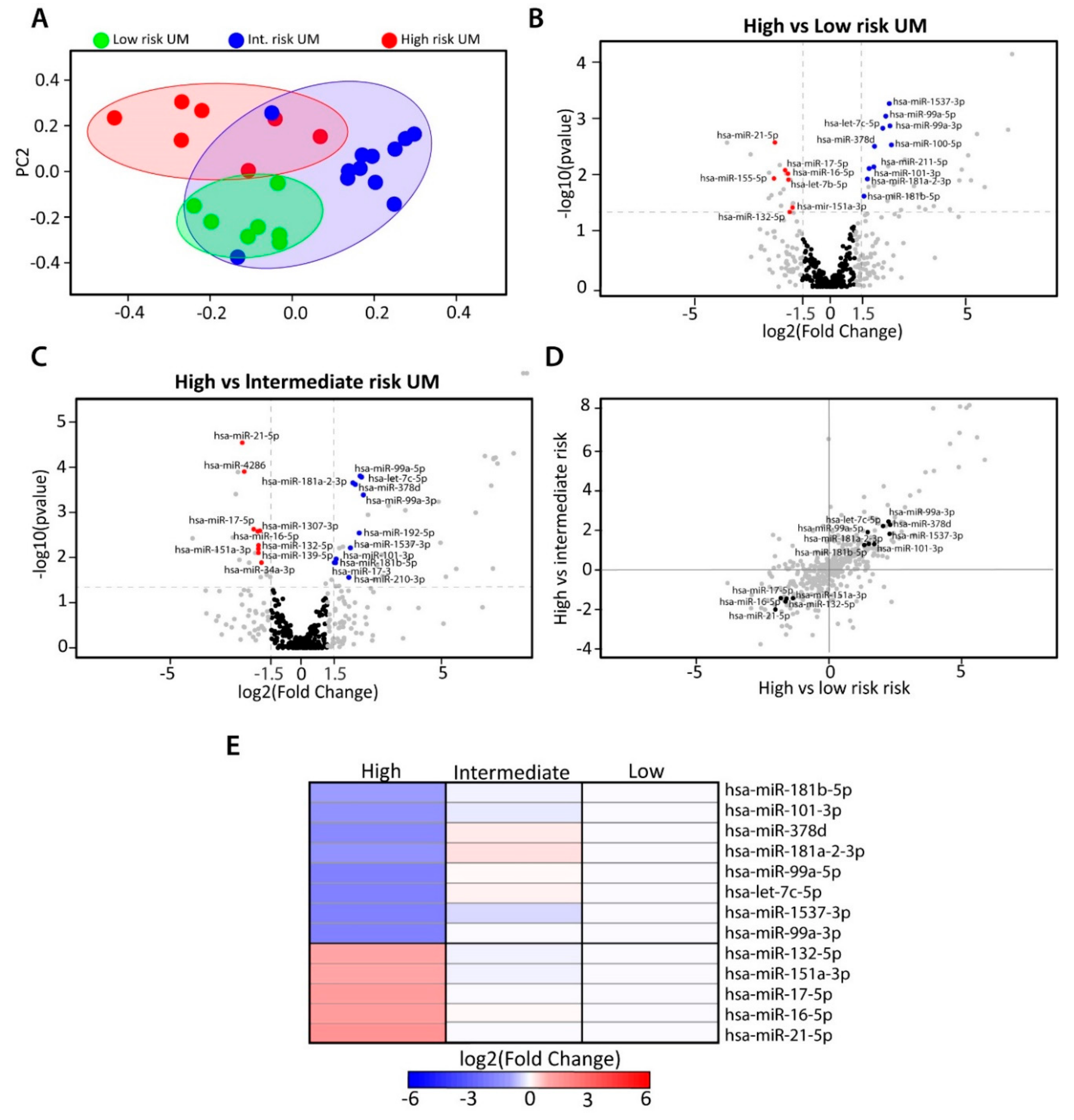
2.2. Identification of Differentially Expressed miRNAs
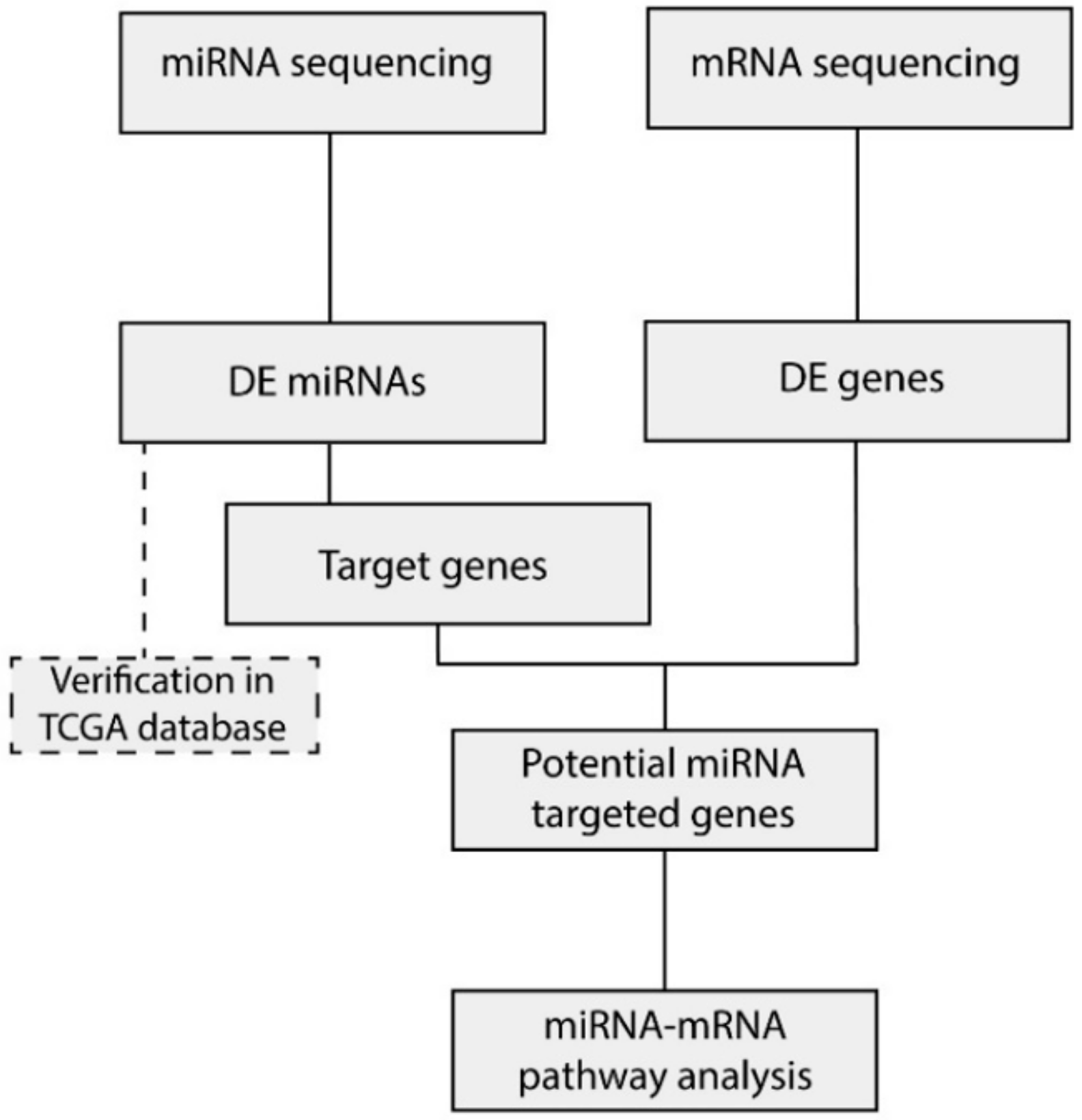
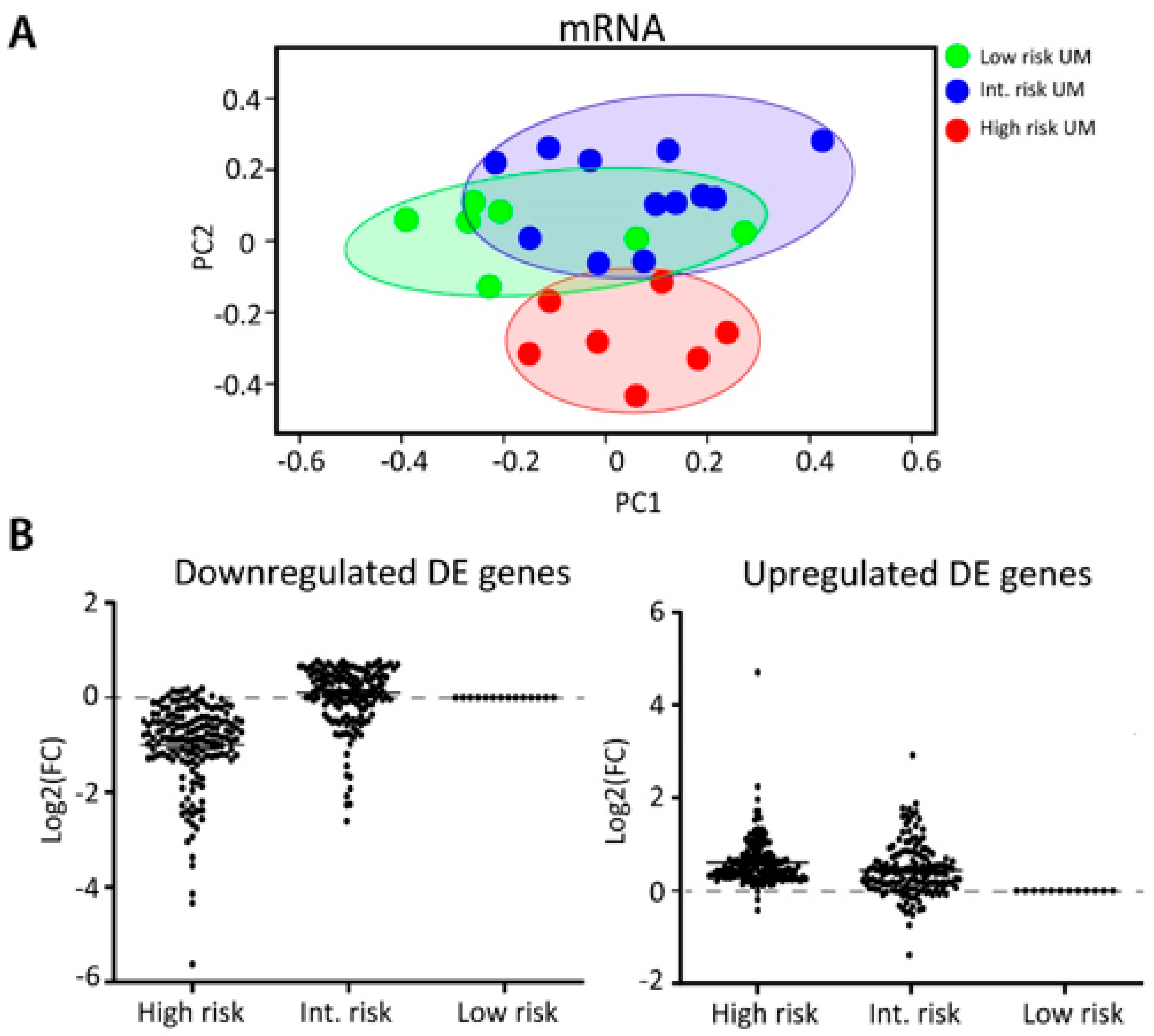
2.3. Integration of miRNA and mRNA Expression Data Identifies Target Genes
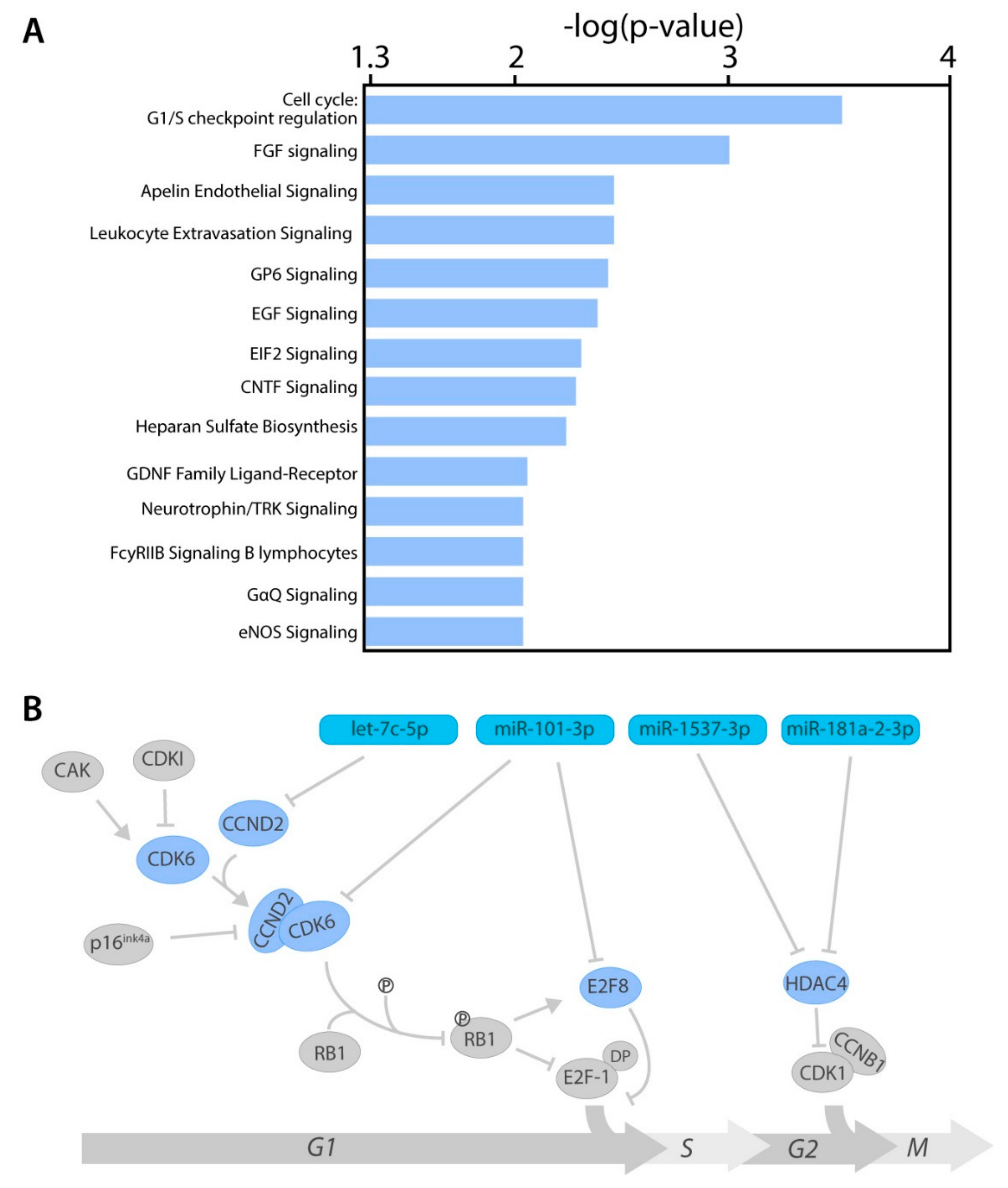
2.4. miRNA Target Genes From Several Cancer-Related Pathways
3. Discussion
4. Materials and Methods
4.1. Tissue Samples
4.2. Mutational Analysis
4.3. Isolation and Sequencing of Small RNA and mRNA
4.4. Analysis of the Sequencing Data
4.5. miRNA Target Gene Prediction and Validation
4.6. Acquisition of TCGA Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Damato, B.; Eleuteri, A.; Taktak, A.F.; Coupland, S.E. Estimating prognosis for survival after treatment of choroidal melanoma. Prog. Retin. Eye Res. 2011, 30, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in gna11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of gnaq in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, A.E.; Vaarwater, J.; Paridaens, D.; Naus, N.C.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study Group. Patient survival in uveal melanoma is not affected by oncogenic mutations in gnaq and gna11. Br. J. Cancer 2013, 109, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study Group. Uveal melanomas with sf3b1 mutations: A distinct subclass associated with late-onset metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of bap1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, N.; Thornton, S.; Coupland, S.E.; Coulson, J.M.; Sacco, J.J.; Krishna, Y.; Heimann, H.; Taktak, A.; Cebulla, C.M.; Abdel-Rahman, M.H.; et al. Patterns of bap1 protein expression provide insights into prognostic significance and the biology of uveal melanoma. J. Pathol. Clin. Res. 2018, 4, 26–38. [Google Scholar] [CrossRef]
- Yu, H.; Mashtalir, N.; Daou, S.; Hammond-Martel, I.; Ross, J.; Sui, G.; Hart, G.W.; Rauscher, F.J.; Drobetsky, E.; Milot, E.; et al. The ubiquitin carboxyl hydrolase bap1 forms a ternary complex with yy1 and hcf-1 and is a critical regulator of gene expression. Mol. Cell Biol. 2010, 30, 5071–5085. [Google Scholar] [CrossRef]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hebert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase bap1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef]
- Martin, M.; Masshofer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in eif1ax and sf3b1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef]
- Harbour, J.W.; Roberson, E.D.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor sf3b1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal micrornas. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Shen, H.; Lu, J.; Li, C.; Hu, D.N.; Dong, X.D.; Yan, D.; Tu, L. Epigenetics, micrornas, and carcinogenesis: Functional role of microrna-137 in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Lou, D. Microrna-34b/c suppresses uveal melanoma cell proliferation and migration through multiple targets. Mol. Vis. 2012, 18, 537–546. [Google Scholar]
- Larsen, A.C.; Holst, L.; Kaczkowski, B.; Andersen, M.T.; Manfe, V.; Siersma, V.D.; Kolko, M.; Kiilgaard, J.F.; Winther, O.; Prause, J.U.; et al. Microrna expression analysis and multiplex ligation-dependent probe amplification in metastatic and non-metastatic uveal melanoma. Acta Ophthalmol. 2014, 92, 541–549. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Shen, J.; Jiang, Y. Microrna dysregulation in uveal melanoma: A new player enters the game. Oncotarget 2015, 6, 4562–4568. [Google Scholar] [CrossRef]
- Liu, J.; Ma, L.; Li, C.; Zhang, Z.; Yang, G.; Zhang, W. Tumor-targeting trail expression mediated by mirna response elements suppressed growth of uveal melanoma cells. Mol. Oncol. 2013, 7, 1043–1055. [Google Scholar] [CrossRef]
- Ma, Y.B.; Song, D.W.; Nie, R.H.; Mu, G.Y. Microrna-32 functions as a tumor suppressor and directly targets ezh2 in uveal melanoma. Genet. Mol. Res. 2016, 15, 7935. [Google Scholar] [CrossRef]
- Ragusa, M.; Barbagallo, C.; Statello, L.; Caltabiano, R.; Russo, A.; Puzzo, L.; Avitabile, T.; Longo, A.; Toro, M.D.; Barbagallo, D.; et al. Mirna profiling in vitreous humor, vitreal exosomes and serum from uveal melanoma patients: Pathological and diagnostic implications. Cancer Biol. Ther. 2015, 16, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Zhou, X.; Chen, X.; Hu, D.N.; Dong, X.D.; Wang, J.; Lu, F.; Tu, L.; Qu, J. Microrna-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-met. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Worley, L.A.; Long, M.D.; Onken, M.D.; Harbour, J.W. Micro-rnas associated with metastasis in uveal melanoma identified by multiplexed microarray profiling. Melanoma Res. 2008, 18, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wei, W. The mirna expression profile of the uveal melanoma. Sci. China Life Sci. 2011, 54, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, H.; Duan, J.; Liu, R.; Deng, T.; Bai, M.; Huang, D.; Li, H.; Ning, T.; Zhang, L.; et al. Mir-17-5p regulates cell proliferation and migration by targeting transforming growth factor-beta receptor 2 in gastric cancer. Oncotarget 2016, 7, 33286–33296. [Google Scholar] [CrossRef]
- Fang, L.; Li, H.; Wang, L.; Hu, J.; Jin, T.; Wang, J.; Yang, B.B. Microrna-17-5p promotes chemotherapeutic drug resistance and tumour metastasis of colorectal cancer by repressing pten expression. Oncotarget 2014, 5, 2974–2987. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhao, H.; Huang, J.; Yan, X.; Zhang, Y.; Gao, Y. Microrna-17-5p promotes gastric cancer proliferation, migration and invasion by directly targeting early growth response 2. Am. J. Cancer Res. 2016, 6, 2010–2020. [Google Scholar] [PubMed]
- Daugaard, I.; Sanders, K.J.; Idica, A.; Vittayarukskul, K.; Hamdorf, M.; Krog, J.D.; Chow, R.; Jury, D.; Hansen, L.L.; Hager, H.; et al. Mir-151a induces partial emt by regulating e-cadherin in nsclc cells. Oncogenesis 2017, 6, e366. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, M.; Guo, T.; Yang, C.; Zhang, C.; Hao, J. Microrna-21-5p promotes proliferation of gastric cancer cells through targeting smad7. Onco. Targets Ther. 2018, 11, 4901–4911. [Google Scholar] [CrossRef]
- Han, M.; Wang, Y.; Liu, M.; Bi, X.; Bao, J.; Zeng, N.; Zhu, Z.; Mo, Z.; Wu, C.; Chen, X. Mir-21 regulates epithelial-mesenchymal transition phenotype and hypoxia-inducible factor-1alpha expression in third-sphere forming breast cancer stem cell-like cells. Cancer Sci. 2012, 103, 1058–1064. [Google Scholar] [CrossRef]
- Cai, L.; Wang, W.; Li, X.; Dong, T.; Zhang, Q.; Zhu, B.; Zhao, H.; Wu, S. Microrna-21-5p induces the metastatic phenotype of human cervical carcinoma cells in vitro by targeting the von hippel-lindau tumor suppressor. Oncol. Lett. 2018, 15, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yang, X.; Wei, W.B.; Xu, X.L. Role of microrna-21 in uveal melanoma cell invasion and metastasis by regulating p53 and its downstream protein. Int. J. Ophthalmol. 2018, 11, 1258–1268. [Google Scholar] [PubMed]
- Rinnerthaler, G.; Hackl, H.; Gampenrieder, S.P.; Hamacher, F.; Hufnagl, C.; Hauser-Kronberger, C.; Zehentmayr, F.; Fastner, G.; Sedlmayer, F.; Mlineritsch, B.; et al. Mir-16-5p is a stably-expressed housekeeping microrna in breast cancer tissues from primary tumors and from metastatic sites. Int. J. Mol. Sci. 2016, 17, 156. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Liu, W. Microrna-99a-5p suppresses breast cancer progression and cell-cycle pathway through downregulating cdc25a. J. Cell Physiol. 2019, 234, 3526–3537. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, B.; Yang, X.; Zhang, C. Mir-99a-5p inhibits bladder cancer cell proliferation by directly targeting mammalian target of rapamycin and predicts patient survival. J. Cell Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shao, M.Y.; Zou, S.C.; Xiao, Z.F.; Chen, Z.C. Mir-101-3p inhibits emt to attenuate proliferation and metastasis in glioblastoma by targeting trim44. J. Neurooncol. 2019, 141, 19–30. [Google Scholar] [CrossRef]
- Zheng, F.; Liao, Y.J.; Cai, M.Y.; Liu, T.H.; Chen, S.P.; Wu, P.H.; Wu, L.; Bian, X.W.; Guan, X.Y.; Zeng, Y.X.; et al. Systemic delivery of microrna-101 potently inhibits hepatocellular carcinoma in vivo by repressing multiple targets. PLoS Genet. 2015, 11, e1004873. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Zheng, X.; Tu, X.; Li, H.; Chen, J.; Zang, Y.; Zhang, J. Loss of microrna-101 promotes epithelial to mesenchymal transition in hepatocytes. J. Cell Physiol. 2015, 230, 2706–2717. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef]
- Tadesse, S.; Yu, M.; Kumarasiri, M.; Le, B.T.; Wang, S. Targeting cdk6 in cancer: State of the art and new insights. Cell Cycle 2015, 14, 3220–3230. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Harbour, J.W. Association between gene expression profile, proliferation and metastasis in uveal melanoma. Curr. Eye Res. 2010, 35, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salas, E.; Pineiro, D.; Fernandez, N. Alternative mechanisms to initiate translation in eukaryotic mrnas. Comp. Funct. Genomics 2012, 2012, 391546. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.K.; Bressler, K.; Patel, H.; Balasingam, N.; Thakor, N. Role of eukaryotic initiation factors during cellular stress and cancer progression. J. Nucleic. Acids 2016, 2016, 8235121. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (egfr) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Korc, M.; Friesel, R.E. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targets 2009, 9, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Hong, X.W.; Wang, L.L.; Cui, X.F.; Lu, J.; Chen, G.Q.; Zheng, Y.L. Microrna-17 inhibition overcomes chemoresistance and suppresses epithelial-mesenchymal transition through a dedd-dependent mechanism in gastric cancer. Int. J. Biochem. Cell Biol. 2018, 102, 59–70. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of micrornas in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating micrornas as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Van Agthoven, T.; Looijenga, L.H.J. Accurate primary germ cell cancer diagnosis using serum based microrna detection (amptsmir test). Oncotarget 2017, 8, 58037–58049. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. Microrna therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microrna function by antimir oligonucleotides. Silence 2012, 3, e1. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Saltzman, W.M.; Slack, F.J. Canonical and non-canonical barriers facing antimir cancer therapeutics. Curr. Med. Chem. 2013, 20, 3582–3593. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, A.E.; Verdijk, R.M.; Brouwer, R.W.; van den Bosch, T.P.; van den Berg, M.M.; Vaarwater, J.; Kockx, C.E.; Paridaens, D.; Naus, N.C.; Nellist, M.; et al. Clinical significance of immunohistochemistry for detection of bap1 mutations in uveal melanoma. Mod. Pathol. 2014, 27, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Derks, K.W.; Misovic, B.; van den Hout, M.C.; Kockx, C.E.; Gomez, C.P.; Brouwer, R.W.; Vrieling, H.; Hoeijmakers, J.H.; van IJcken, W.F.; Pothof, J. Deciphering the rna landscape by rnaome sequencing. RNA Biol. 2015, 12, 30–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. Stem: A tool for the analysis of short time series gene expression data. BMC Bioinf. 2006, 7, e191. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. Mirwalk: An online resource for prediction of microrna binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microrna target sites in mammalian mrnas. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Maragkakis, M.; Reczko, M.; Simossis, V.A.; Alexiou, P.; Papadopoulos, G.L.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; et al. Diana-microt web server: Elucidating microrna functions through target prediction. Nucleic. Acids Res. 2009, 37, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Wang, X. Mirdb: An online resource for microrna target prediction and functional annotations. Nucleic. Acids Res. 2015, 43, 146–152. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients Characteristics | Low Risk Group | Intermediate Risk Group | High Risk Group |
|---|---|---|---|
| (n = 7) | (n = 12) | (n = 7) | |
| Age | |||
| Mean ± SD | 58 ± 9 | 50 ± 15 | 69 ± 14 |
| Gender, N (%) | |||
| Male | 5 (71) | 5 (42) | 1 (14) |
| Female | 2 (29) | 7 (58) | 6 (86) |
| Disease free survival | |||
| Mean ± SD | 145.1 ± 45.1 | 103.3 ± 50.6 | 28.2 ± 9.26 |
| Mutation status, N (%) | |||
| GNAQ | 4 (57) | 7 (58) | 4 (57) |
| GAN11 | 3 (43) | 5 (42) | 3 (43) |
| EIF1AX | 7 (100) | 0 (0) | 0 (0) |
| SF3B1 | 0 (0) | 12 (100) | 0 (0) |
| BAP1 | 0 (0) | 0 (0) | 7 (100) |
| Monosomy 3, N (%) | |||
| Present | 0 (0) | 0 (0) | 7 (100) |
| Absent | 7 (100) | 11 (92) | 0 (0) |
| NE | 0(0) | 1 (8) | 0 (0) |
| BAP1 IHC, N (%) | |||
| Positive | 7 (100) | 12 (100) | 0 (0) |
| Negative | 0 (0) | 0 (0) | 7 (100) |
| Metastasis | |||
| Present | 0 (0) | 9 (75) | 7 (100) |
| Absent | 7 (0) | 3 (25) | 0 (0) |
| miRNA | Target Gene |
|---|---|
| let-7c-5p | ACSL6, AGO4, CACNB4, CCND2, CUX1, ESPL1, FRMD4B, LINGO1, MTDH, PALD1, PARP8, RDX, RGS16 *, RNF217, STARD13 |
| miR-16-5p | CNTN3, COL24A1, DIXDC1, ESRRG, EXTL3, FAM110C, FGF2, HS3ST5, ITPR1, MBNL2, OTUD4, PDK4, SLC6A11, SLC7A2, SNRK, SOX5 *, SYT3, VEGFA, ZMAT3 |
| miR-17-5p | ARAP2, CDC5L, DCBLD2, ENPP5, ETV1, HMGB3, NR4A3, NTNG1, PCDHA6, SESN3, SLC12A3, TUSC2 |
| miR-21-5p | AMER1 *, BCL11A, CSRNP3, IRAK1BP1, LIFR, MEF2C, NKIRAS1, PAIP2B, PCSK6, PDZD2, PRPF4B, STATB1, SCN8A *, SLC22A15, SPRY1, ST6GAL, TIMP3 |
| miR-99a-5p | FGFR3, HS3STB1 |
| miR-99a-3p | GIMAP1 |
| miR-101-3p | ASAP1, ATAD2B, C8orf44-SGK3, CDK6, CLDN11, E2F8, HSD11B2, IMPA1, ITGA8, MAGI2 *, MYCN, PBX3 *, PHACTR2, SALL1, SGK3, SH3PXD2A, SORL1, SRGAP *, STOX2, TRIB1, TSHZ3, ZDHHC21 |
| miR-132-5p | ANO10, DUSP7 |
| miR-151a-3p | SEC22C |
| miR-181a-2-3p | HDAC4 *, PAG1, RBMS3, ZFP62 |
| miR-181b-5p | DERL1, FBXL17, MBOAT2, PLAG, SLC7A, YTHDF3 * |
| miR-378d | PHF21B, PTPN11 |
| miR-1537-3p | NSMAF, SNX8, SPC25, TNFSF15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smit, K.N.; Chang, J.; Derks, K.; Vaarwater, J.; Brands, T.; Verdijk, R.M.; Wiemer, E.A.C.; Mensink, H.W.; Pothof, J.; de Klein, A.; et al. Aberrant MicroRNA Expression and Its Implications for Uveal Melanoma Metastasis. Cancers 2019, 11, 815. https://doi.org/10.3390/cancers11060815
Smit KN, Chang J, Derks K, Vaarwater J, Brands T, Verdijk RM, Wiemer EAC, Mensink HW, Pothof J, de Klein A, et al. Aberrant MicroRNA Expression and Its Implications for Uveal Melanoma Metastasis. Cancers. 2019; 11(6):815. https://doi.org/10.3390/cancers11060815
Chicago/Turabian StyleSmit, Kyra N., Jiang Chang, Kasper Derks, Jolanda Vaarwater, Tom Brands, Rob M. Verdijk, Erik A.C. Wiemer, Hanneke W. Mensink, Joris Pothof, Annelies de Klein, and et al. 2019. "Aberrant MicroRNA Expression and Its Implications for Uveal Melanoma Metastasis" Cancers 11, no. 6: 815. https://doi.org/10.3390/cancers11060815
APA StyleSmit, K. N., Chang, J., Derks, K., Vaarwater, J., Brands, T., Verdijk, R. M., Wiemer, E. A. C., Mensink, H. W., Pothof, J., de Klein, A., & Kilic, E. (2019). Aberrant MicroRNA Expression and Its Implications for Uveal Melanoma Metastasis. Cancers, 11(6), 815. https://doi.org/10.3390/cancers11060815