Contribution of Mitochondrial Ion Channels to Chemo-Resistance in Cancer Cells


Abstract
1. Introduction
2. Defective Mitochondrial Outer Membrane Permeabilization as a Cause of Chemo-Resistance
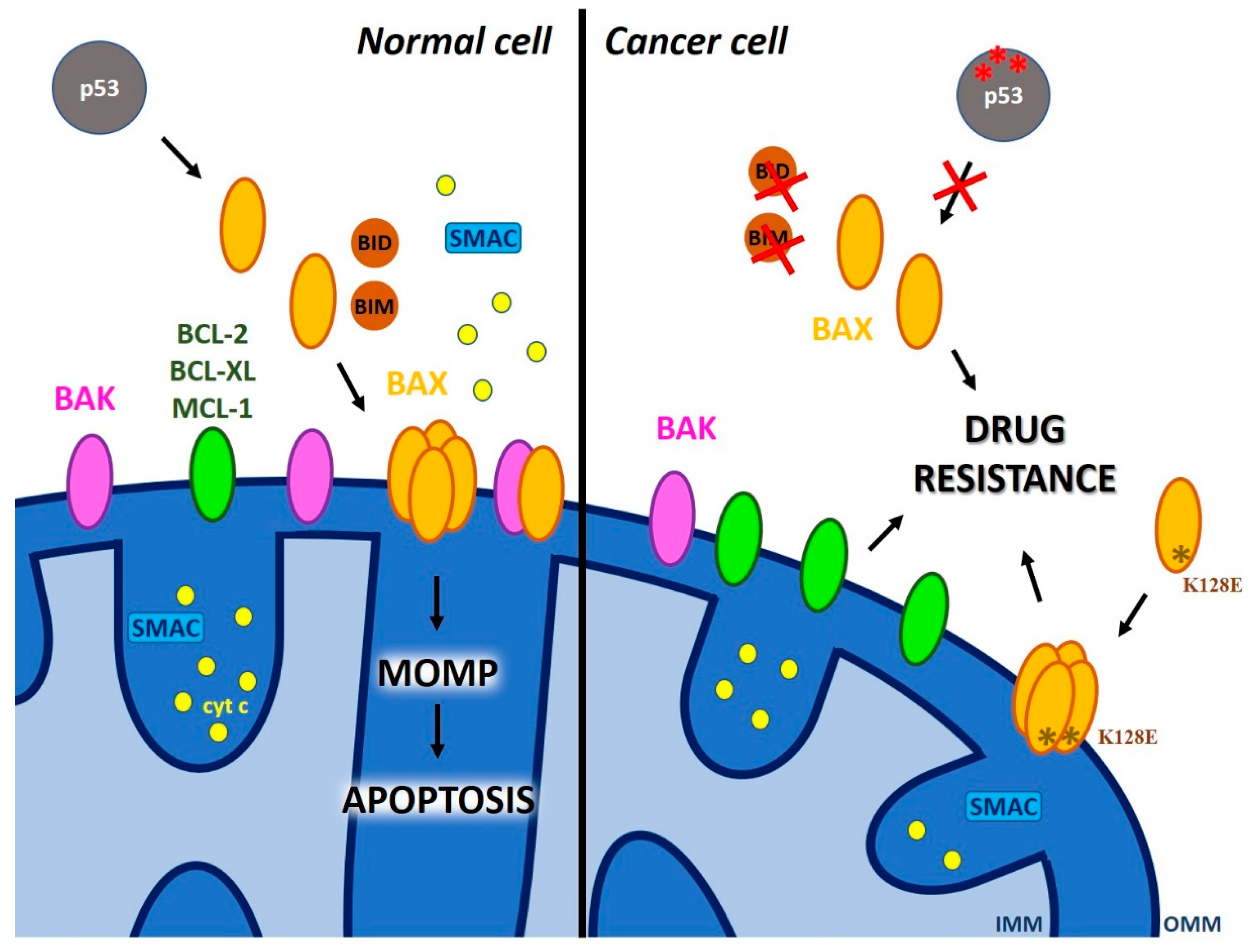
2.1. The Role of Pore-Forming Pro-Apoptotic MOM Proteins of the BCL-2 Family in MOMP
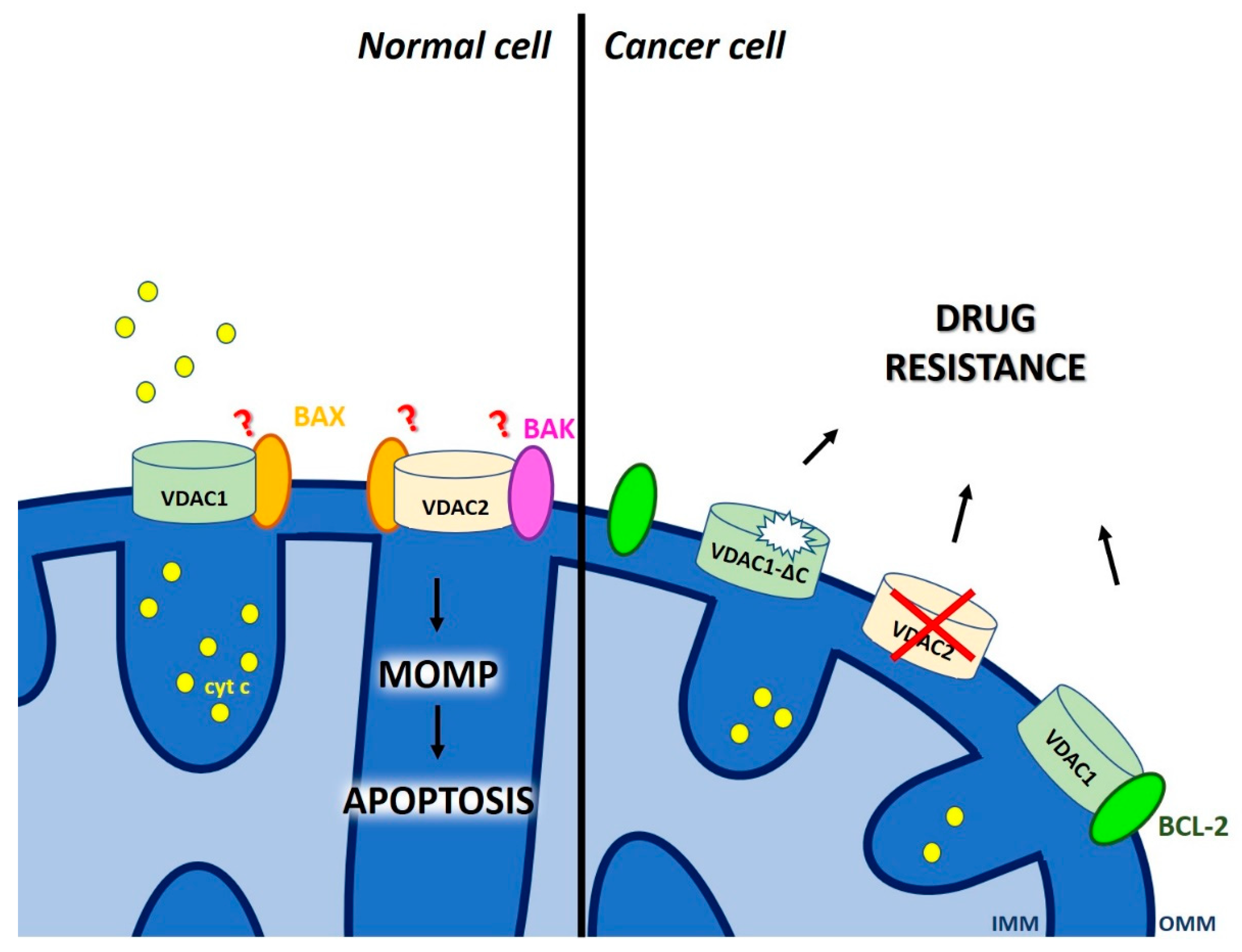
2.2. Dual Role of Voltage-Dependent Anion Channels (VDAC) in Chemo-Resistance
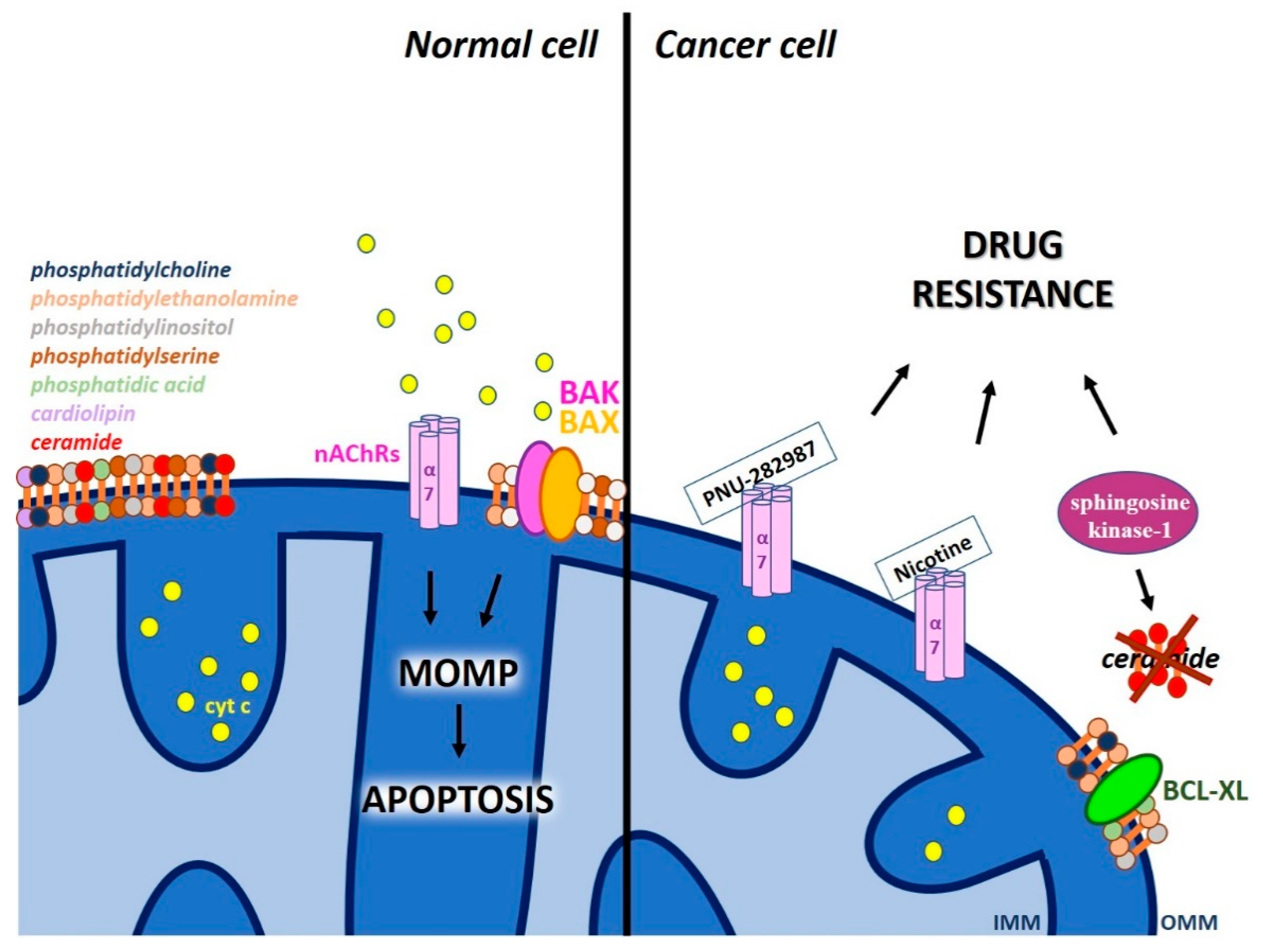
2.3. Other Proteic Channels and Lipids of The MOM that Modulate MOMP
3. Defective Mitochondrial Inner Membrane Permeabilization Leading to Chemo-Resistance
3.1. The Mitochondrial Permeability Transition
3.2. Calcium Channels in the Inner Mitochondrial Membrane Linked to Chemo-Resistance
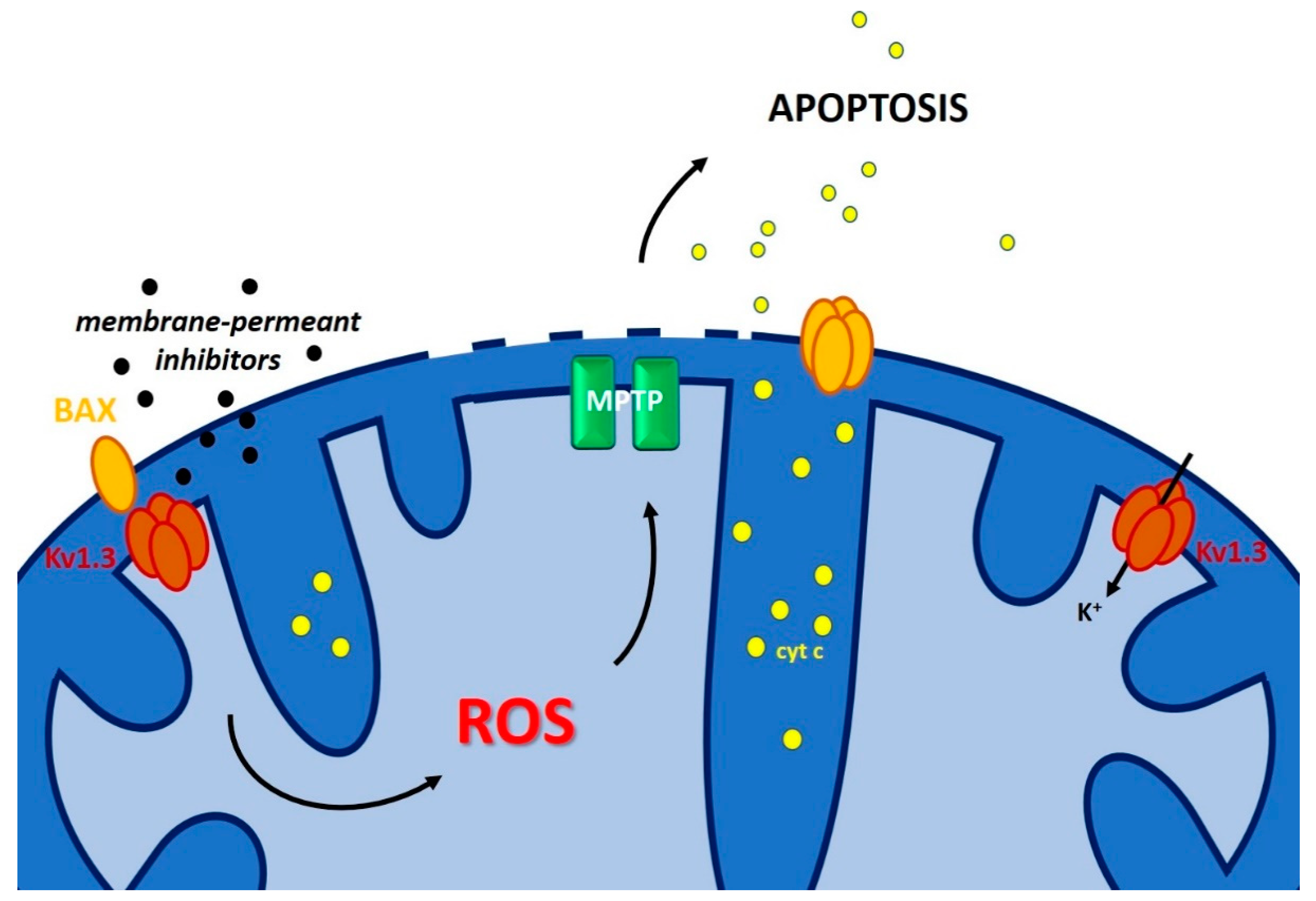
3.3. Inner Membrane Potassium Channels and Chemo-Resistance
3.4. Other Channels
4. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondrial regulation of cell death: A phylogenetically conserved control. Microb. Cell (Graz Austria) 2016, 3, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Chen, J.; Bi, X.; Li, Z.; Gao, X.; Li, H.; Zhu, H.; Huang, Y.; Qi, J.; Zhang, Y. Overcoming Multidrug Resistance through the GLUT1-Mediated and Enzyme-Triggered Mitochondrial Targeting Conjugate with Redox-Sensitive Paclitaxel Release. ACS Appl. Mater. Interfaces 2018, 10, 12351–12363. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.F.; Rong, W.T.; Lu, Y.; Hou, J.; Qi, S.S.; Xiao, Q.; Zhang, J.; You, J.; Yu, S.Q.; Xu, Q. TPGS2k/PLGA nanoparticles for overcoming multidrug resistance by interfering mitochondria of human alveolar adenocarcinoma cells. ACS Appl. Mater. Interfaces 2015, 7, 3888–3901. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kroemer, G. Organelle-specific initiation of cell death. Nat. Cell Biol. 2014, 16, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta 2016, 1857, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 2014, 56, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Fieni, F.; Parkar, A.; Misgeld, T.; Kerschensteiner, M.; Lichtman, J.W.; Pasinelli, P.; Trotti, D. Voltage-dependent inwardly rectifying potassium conductance in the outer membrane of neuronal mitochondria. J. Biol. Chem. 2010, 285, 27411–27417. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Stefani, E.; Toro, L. Intracellular BK(Ca) (iBK(Ca)) channels. J. Physiol. 2012, 590, 5937–5947. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabo, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009, 45, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Netter, M.F.; Perocchi, F.; Doti, N.; Meissner, L.; Tobaben, S.; Grohm, J.; Zischka, H.; Plesnila, N.; Decher, N.; et al. Mitochondrial small conductance SK2 channels prevent glutamate-induced oxytosis and mitochondrial dysfunction. J. Biol. Chem. 2013, 288, 10792–10804. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Bock, J.; Jekle, A.; Soddemann, M.; Adams, C.; Lang, F.; Zoratti, M.; Gulbins, E. A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 2005, 280, 12790–12798. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Zoratti, M.; Gulbins, E.; Szabo, I. Induction of apoptosis in macrophages via Kv1.3 and Kv1.5 potassium channels. Curr. Med. Chem. 2012, 19, 5394–5404. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Barrese, V.; Soldovieri, M.V.; Ambrosino, P.; Martelli, A.; Vinciguerra, I.; Miceli, F.; Greenwood, I.; Curtis, M.J.; Breschi, M.C.; et al. Expression and function of Kv7.4 channels in Rat cardiac mitochondria: Possible targets for cardioprotection. Cardiovasc. Res. 2015, 110, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef]
- Pocsai, K.; Kosztka, L.; Bakondi, G.; Gonczi, M.; Fodor, J.; Dienes, B.; Szentesi, P.; Kovacs, I.; Feniger-Barish, R.; Kopf, E.; et al. Melanoma cells exhibit strong intracellular TASK-3-specific immunopositivity in both tissue sections and cell culture. Cell. Mol. Life Sci. CMLS 2006, 63, 2364–2376. [Google Scholar] [CrossRef]
- Reed, J.C. Apoptosis-based therapies. Nat. Rev. Drug Discov. 2002, 1, 111–121. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Pena-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond - mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Cline, K. Mechanistic Aspects of Folded Protein Transport by the Twin Arginine Translocase (Tat). J. Biol. Chem. 2015, 290, 16530–16538. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Rouse, S.L.; Tait, C.E.; Harmer, J.; De Riso, A.; Timmel, C.R.; Sansom, M.S.; Berks, B.C.; Schnell, J.R. Structural model for the protein-translocating element of the twin-arginine transport system. Proc. Natl. Acad. Sci. USA 2013, 110, E1092–E1101. [Google Scholar] [CrossRef]
- Antonsson, B.; Conti, F.; Ciavatta, A.; Montessuit, S.; Lewis, S.; Martinou, I.; Bernasconi, L.; Bernard, A.; Mermod, J.J.; Mazzei, G.; et al. Inhibition of Bax channel-forming activity by Bcl-2. Science (New York N.Y.) 1997, 277, 370–372. [Google Scholar] [CrossRef]
- Hetz, C.; Vitte, P.A.; Bombrun, A.; Rostovtseva, T.K.; Montessuit, S.; Hiver, A.; Schwarz, M.K.; Church, D.J.; Korsmeyer, S.J.; Martinou, J.C.; et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J. Biol. Chem. 2005, 280, 42960–42970. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Antonsson, B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis 2007, 12, 857–868. [Google Scholar] [CrossRef]
- Peixoto, P.M.; Teijido, O.; Mirzalieva, O.; Dejean, L.M.; Pavlov, E.V.; Antonsson, B.; Kinnally, K.W. MAC inhibitors antagonize the pro-apoptotic effects of tBid and disassemble Bax/Bak oligomers. J. Bioenerg. Biomembr. 2017, 49, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Annis, M.G.; Soucie, E.L.; Dlugosz, P.J.; Cruz-Aguado, J.A.; Penn, L.Z.; Leber, B.; Andrews, D.W. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005, 24, 2096–2103. [Google Scholar] [CrossRef] [PubMed]
- Westphal, D.; Dewson, G.; Menard, M.; Frederick, P.; Iyer, S.; Bartolo, R.; Gibson, L.; Czabotar, P.E.; Smith, B.J.; Adams, J.M.; et al. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, E4076–E4085. [Google Scholar] [CrossRef] [PubMed]
- Precht, T.A.; Phelps, R.A.; Linseman, D.A.; Butts, B.D.; Le, S.S.; Laessig, T.A.; Bouchard, R.J.; Heidenreich, K.A. The permeability transition pore triggers Bax translocation to mitochondria during neuronal apoptosis. Cell Death Differ. 2005, 12, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Soddemann, M.; Leanza, L.; Zoratti, M.; Gulbins, E. Single-point mutations of a lysine residue change function of Bax and Bcl-xL expressed in Bax- and Bak-less mouse embryonic fibroblasts: Novel insights into the molecular mechanisms of Bax-induced apoptosis. Cell Death Differ. 2011, 18, 427–438. [Google Scholar] [CrossRef]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; Garcia-Saez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef]
- Grosse, L.; Wurm, C.A.; Bruser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016, 35, 402–413. [Google Scholar] [CrossRef]
- Nasu, Y.; Benke, A.; Arakawa, S.; Yoshida, G.J.; Kawamura, G.; Manley, S.; Shimizu, S.; Ozawa, T. In Situ Characterization of Bak Clusters Responsible for Cell Death Using Single Molecule Localization Microscopy. Sci. Rep. 2016, 6, 27505. [Google Scholar] [CrossRef]
- Bove, J.; Martinez-Vicente, M.; Dehay, B.; Perier, C.; Recasens, A.; Bombrun, A.; Antonsson, B.; Vila, M. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 2014, 10, 889–900. [Google Scholar] [CrossRef]
- Kasahara, A.; Scorrano, L. Mitochondria: From cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef]
- Anderson, M.A.; Huang, D.; Roberts, A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin. Hematol. 2014, 51, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Touzeau, C.; Maciag, P.; Amiot, M.; Moreau, P. Targeting Bcl-2 for the treatment of multiple myeloma. Leukemia 2018, 32, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Levis, M.J. Advances in targeted therapy for acute myeloid leukaemia. Br. J. Haematol. 2018, 180, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Slone, S.R.; Lesort, M.; Yacoubian, T.A. 14-3-3theta protects against neurotoxicity in a cellular Parkinson’s disease model through inhibition of the apoptotic factor Bax. PLoS ONE 2011, 6, e21720. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Valentijn, A.J.; Zhang, L.; Gilmore, A.P. The N-terminal conformation of Bax regulates cell commitment to apoptosis. Cell Death Differ. 2007, 14, 932–942. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef]
- Martel, C.; Wang, Z.; Brenner, C. VDAC phosphorylation, a lipid sensor influencing the cell fate. Mitochondrion 2014, 19 Pt A, 69–77. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Chen, Q. VDAC1 as a Player in Mitochondria-Mediated Apoptosis and Target for Modulating Apoptosis. Curr. Med. Chem. 2017, 24, 4435–4446. [Google Scholar] [CrossRef]
- Becker, T.; Wagner, R. Mitochondrial Outer Membrane Channels: Emerging Diversity in Transport Processes. BioEssays 2018, 40, e1800013. [Google Scholar] [CrossRef] [PubMed]
- Checchetto, V.; Szabo, I. Novel Channels of the Outer Membrane of Mitochondria: Recent Discoveries Change Our View. BioEssays 2018, 40, e1700232. [Google Scholar] [CrossRef] [PubMed]
- Kruger, V.; Becker, T.; Becker, L.; Montilla-Martinez, M.; Ellenrieder, L.; Vogtle, F.N.; Meyer, H.E.; Ryan, M.T.; Wiedemann, N.; Warscheid, B.; et al. Identification of new channels by systematic analysis of the mitochondrial outer membrane. J. Cell Biol. 2017, 216, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. The Warburg effect and mitochondrial stability in cancer cells. Mol. Asp. Med. 2010, 31, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.M. Targeting mitochondria for cancer therapy. Environ. Mol Mutagen. 2010, 51, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kroemer, G. Mitochondria as therapeutic targets for the treatment of malignant disease. Antioxid. Redox Signal. 2011, 15, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Pahima, H.; Reina, S.; Tadmor, N.; Dadon-Klein, D.; Shteinfer-Kuzmine, A.; Mazure, N.M.; De Pinto, V.; Shoshan-Barmatz, V. Hypoxic-induced truncation of voltage-dependent anion channel 1 is mediated by both asparagine endopeptidase and calpain 1 activities. Oncotarget 2018, 9, 12825–12841. [Google Scholar] [CrossRef][Green Version]
- Brahimi-Horn, M.C.; Ben-Hail, D.; Ilie, M.; Gounon, P.; Rouleau, M.; Hofman, V.; Doyen, J.; Mari, B.; Shoshan-Barmatz, V.; Hofman, P.; et al. Expression of a truncated active form of VDAC1 in lung cancer associates with hypoxic cell survival and correlates with progression to chemotherapy resistance. Cancer Res. 2012, 72, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Ferecatu, I.; Canal, F.; Fabbri, L.; Mazure, N.M.; Bouton, C.; Golinelli-Cohen, M.P. Dysfunction in the mitochondrial Fe-S assembly machinery leads to formation of the chemoresistant truncated VDAC1 isoform without HIF-1alpha activation. PLoS ONE 2018, 13, e0194782. [Google Scholar] [CrossRef]
- Dehghan-Nayeri, N.; Rezaei-Tavirani, M.; Omrani, M.D.; Gharehbaghian, A.; Goudarzi Pour, K.; Eshghi, P. Identification of potential predictive markers of dexamethasone resistance in childhood acute lymphoblastic leukemia. J. Cell Commun. Signal. 2017, 11, 137–145. [Google Scholar] [CrossRef]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [PubMed]
- Zalk, R.; Israelson, A.; Garty, E.S.; Azoulay-Zohar, H.; Shoshan-Barmatz, V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 2005, 386, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Bergdoll, L.A.; Lerch, M.T.; Patrick, J.W.; Belardo, K.; Altenbach, C.; Bisignano, P.; Laganowsky, A.; Grabe, M.; Hubbell, W.L.; Abramson, J. Protonation state of glutamate 73 regulates the formation of a specific dimeric association of mVDAC1. Proc. Natl. Acad. Sci. USA 2018, 115, E172–E179. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Okazaki, M.; Kimura-Someya, T.; Ishizuka-Katsura, Y.; Ito, K.; Yokoyama, S.; Dodo, K.; Sodeoka, M.; Shirouzu, M. Crystal structural characterization reveals novel oligomeric interactions of human voltage-dependent anion channel 1. Protein Soc. 2017, 26, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Ide, T.; Yanagida, T.; Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000, 275, 12321–12325. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Han, J.; Ben-Hail, D.; He, L.; Li, B.; Chen, Z.; Wang, Y.; Yang, Y.; Liu, L.; Zhu, Y.; et al. A New Fungal Diterpene Induces VDAC1-dependent Apoptosis in Bax/Bak-deficient Cells. J. Biol. Chem. 2015, 290, 23563–23578. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science (New York N.Y.) 2003, 301, 513–517. [Google Scholar] [CrossRef]
- Ma, S.B.; Nguyen, T.N.; Tan, I.; Ninnis, R.; Iyer, S.; Stroud, D.A.; Menard, M.; Kluck, R.M.; Ryan, M.T.; Dewson, G. Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: A requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ. 2014, 21, 1925–1935. [Google Scholar] [CrossRef]
- Yamagata, H.; Shimizu, S.; Nishida, Y.; Watanabe, Y.; Craigen, W.J.; Tsujimoto, Y. Requirement of voltage-dependent anion channel 2 for pro-apoptotic activity of Bax. Oncogene 2009, 28, 3563–3572. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.S.; Li, M.X.; Tan, I.K.L.; Ninnis, R.L.; Reljic, B.; Scicluna, K.; Dagley, L.F.; Sandow, J.J.; Kelly, G.L.; Samson, A.L.; et al. VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nat. Commun. 2018, 9, 4976. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Fiorillo, M.; Lisanti, M.P. Mitochondrial markers predict recurrence, metastasis and tamoxifen-resistance in breast cancer patients: Early detection of treatment failure with companion diagnostics. Oncotarget 2017, 8, 68730–68745. [Google Scholar] [CrossRef] [PubMed]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Prezma, T.; Shteinfer, A.; Admoni, L.; Raviv, Z.; Sela, I.; Levi, I.; Shoshan-Barmatz, V. VDAC1-based peptides: Novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis. 2013, 4, e809. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shteinfer-Kuzmine, A.; Amsalem, Z.; Arif, T.; Zooravlov, A.; Shoshan-Barmatz, V. Selective induction of cancer cell death by VDAC1-based peptides and their potential use in cancer therapy. Mol. Oncol. 2018, 12, 1077–1103. [Google Scholar] [CrossRef] [PubMed]
- Pittala, S.; Krelin, Y.; Shoshan-Barmatz, V. Targeting Liver Cancer and Associated Pathologies in Mice with a Mitochondrial VDAC1-Based Peptide. Neoplasia (New York N.Y.) 2018, 20, 594–609. [Google Scholar] [CrossRef]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef]
- Leanza, L.; Checchetto, V.; Biasutto, L.; Rossa, A.; Costa, R.; Bachmann, M.; Zoratti, M.; Szabo, I. Pharmacological modulation of mitochondrial ion channels. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Checchetto, V.; Reina, S.; Magri, A.; Szabo, I.; De Pinto, V. Recombinant human voltage dependent anion selective channel isoform 3 (hVDAC3) forms pores with a very small conductance. Cell. Physiol. Biochem. 2014, 34, 842–853. [Google Scholar] [CrossRef]
- Reina, S.; Guarino, F.; Magri, A.; De Pinto, V. VDAC3 As a Potential Marker of Mitochondrial Status Is Involved in Cancer and Pathology. Front. Oncol. 2016, 6, 264. [Google Scholar] [CrossRef] [PubMed]
- Kalashnyk, O.M.; Gergalova, G.L.; Komisarenko, S.V.; Skok, M.V. Intracellular localization of nicotinic acetylcholine receptors in human cell lines. Life Sci. 2012, 91, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.I.; Shchepotin, I.B.; Galitovkiy, V.; Grando, S.A. Mechanisms of tumor-promoting activities of nicotine in lung cancer: Synergistic effects of cell membrane and mitochondrial nicotinic acetylcholine receptors. BMC Cancer 2015, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Huang, C.Y.; Cheng, W.L.; Hung, C.S.; Huang, M.T.; Tai, C.J.; Liu, Y.N.; Chen, C.L.; Chang, Y.J. Alpha 7-nicotinic acetylcholine receptor mediates the sensitivity of gastric cancer cells to 5-fluorouracil. Tumour Biol. 2015, 36, 9537–9544. [Google Scholar] [CrossRef]
- Tu, C.C.; Huang, C.Y.; Cheng, W.L.; Hung, C.S.; Uyanga, B.; Wei, P.L.; Chang, Y.J. The alpha7-nicotinic acetylcholine receptor mediates the sensitivity of gastric cancer cells to taxanes. Tumour Biol. 2016, 37, 4421–4428. [Google Scholar] [CrossRef]
- Tu, C.C.; Huang, C.Y.; Cheng, W.L.; Hung, C.S.; Chang, Y.J.; Wei, P.L. Silencing A7-nAChR levels increases the sensitivity of gastric cancer cells to ixabepilone treatment. Tumour Biol. 2016, 37, 9493–9501. [Google Scholar] [CrossRef]
- Uspenska, K.; Lykhmus, O.; Obolenskaya, M.; Pons, S.; Maskos, U.; Komisarenko, S.; Skok, M. Mitochondrial Nicotinic Acetylcholine Receptors Support Liver Cells Viability After Partial Hepatectomy. Front. Pharmacol. 2018, 9, 626. [Google Scholar] [CrossRef]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express alpha7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: Study on isolated mitochondria. PLoS ONE 2012, 7, e31361. [Google Scholar] [CrossRef]
- Trevino, J.G.; Pillai, S.; Kunigal, S.; Singh, S.; Fulp, W.J.; Centeno, B.A.; Chellappan, S.P. Nicotine induces inhibitor of differentiation-1 in a Src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia (New York N.Y.) 2012, 14, 1102–1114. [Google Scholar] [CrossRef]
- Chernyavsky, A.I.; Shchepotin, I.B.; Grando, S.A. Mechanisms of growth-promoting and tumor-protecting effects of epithelial nicotinic acetylcholine receptors. Int. Immunopharmacol. 2015, 29, 36–44. [Google Scholar] [CrossRef]
- Singh, S.; Pillai, S.; Chellappan, S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J. Oncol. 2011, 2011, 456743. [Google Scholar] [CrossRef] [PubMed]
- Dinicola, S.; Morini, V.; Coluccia, P.; Proietti, S.; D’Anselmi, F.; Pasqualato, A.; Masiello, M.G.; Palombo, A.; De Toma, G.; Bizzarri, M.; et al. Nicotine increases survival in human colon cancer cells treated with chemotherapeutic drugs. Toxicology In Vitro 2013, 27, 2256–2263. [Google Scholar] [CrossRef] [PubMed]
- Renault, T.T.; Chipuk, J.E. Death upon a kiss: Mitochondrial outer membrane composition and organelle communication govern sensitivity to BAK/BAX-dependent apoptosis. Chem. Biol. 2014, 21, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R.; Newmeyer, D.D. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef]
- Schafer, B.; Quispe, J.; Choudhary, V.; Chipuk, J.E.; Ajero, T.G.; Du, H.; Schneiter, R.; Kuwana, T. Mitochondrial outer membrane proteins assist Bid in Bax-mediated lipidic pore formation. Mol. Biol. Cell 2009, 20, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Iverson, S.L.; Enoksson, M.; Gogvadze, V.; Ott, M.; Orrenius, S. Cardiolipin is not required for Bax-mediated cytochrome c release from yeast mitochondria. J. Biol. Chem. 2004, 279, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, L.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef]
- Colombini, M. Ceramide channels and mitochondrial outer membrane permeability. J. Bioenerg. Biomembr. 2017, 49, 57–64. [Google Scholar] [CrossRef]
- Chang, K.T.; Anishkin, A.; Patwardhan, G.A.; Beverly, L.J.; Siskind, L.J.; Colombini, M. Ceramide channels: Destabilization by Bcl-xL and role in apoptosis. Biochim. Biophys. Acta 2015, 1848, 2374–2384. [Google Scholar] [CrossRef]
- Bonhoure, E.; Pchejetski, D.; Aouali, N.; Morjani, H.; Levade, T.; Kohama, T.; Cuvillier, O. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia 2006, 20, 95–102. [Google Scholar] [CrossRef]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Muller, D.; Schneider, M.; et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc. Res. 2005, 66, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360. [Google Scholar] [PubMed]
- Szabo, I.; Zoratti, M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J. Biol. Chem. 1991, 266, 3376–3379. [Google Scholar] [PubMed]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabo, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [PubMed]
- Szabo, I.; Zoratti, M. The mitochondrial megachannel is the permeability transition pore. J. Bioenerg. Biomembr. 1992, 24, 111–117. [Google Scholar] [CrossRef]
- Szabo, I.; Bernardi, P.; Zoratti, M. Modulation of the mitochondrial megachannel by divalent cations and protons. J. Biol. Chem. 1992, 267, 2940–2946. [Google Scholar] [PubMed]
- Morisaki, T.; Katano, M. Mitochondria-targeting therapeutic strategies for overcoming chemoresistance and progression of cancer. Curr. Med. Chem. 2003, 10, 2517–2521. [Google Scholar] [CrossRef]
- Leanza, L.; Zoratti, M.; Gulbins, E.; Szabo, I. Mitochondrial ion channels as oncological targets. Oncogene 2014, 33, 5569–5581. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Biasutto, L.; Azzolini, M.; Szabo, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta 2016, 1863, 2515–2530. [Google Scholar] [CrossRef]
- Zhang, R.; Li, G.; Zhang, Q.; Tang, Q.; Huang, J.; Hu, C.; Liu, Y.; Wang, Q.; Liu, W.; Gao, N.; et al. Hirsutine induces mPTP-dependent apoptosis through ROCK1/PTEN/PI3K/GSK3beta pathway in human lung cancer cells. Cell Death Dis. 2018, 9, 598. [Google Scholar] [CrossRef] [PubMed]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic acid induces cytochrome c release and apoptosis in a Bax/Bak-independent, permeability transition pore dependent fashion. Apoptosis 2009, 14, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Cerimele, F.; Ushio-Fukai, M.; Waqas, M.; Campbell, P.M.; Govindarajan, B.; Der, C.J.; Battle, T.; Frank, D.A.; Ye, K.; et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem. 2003, 278, 35501–35507. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Hideshima, T.; Hamasaki, M.; Raje, N.; Kumar, S.; Hideshima, H.; Shiraishi, N.; Yasui, H.; Roccaro, A.M.; Richardson, P.; et al. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood 2005, 106, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef]
- Carraro, M.; Checchetto, V.; Sartori, G.; Kucharczyk, R.; di Rago, J.P.; Minervini, G.; Franchin, C.; Arrigoni, G.; Giorgio, V.; Petronilli, V.; et al. High-Conductance Channel Formation in Yeast Mitochondria is Mediated by F-ATP Synthase e and g Subunits. Cell. Physiol. Biochem. 2018, 50, 1840–1855. [Google Scholar] [CrossRef]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17.e12. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; De Bortoli, S.; Schwarzlander, M.; Szabo, I. Regulation of mitochondrial calcium in plants versus animals. J. Exp. Bot. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pendin, D.; Greotti, E.; Pozzan, T. The elusive importance of being a mitochondrial Ca(2+) uniporter. Cell Calcium 2014, 55, 139–145. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef]
- Antony, A.N.; Paillard, M.; Moffat, C.; Juskeviciute, E.; Correnti, J.; Bolon, B.; Rubin, E.; Csordas, G.; Seifert, E.L.; Hoek, J.B.; et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat. Commun. 2016, 7, 10955. [Google Scholar] [CrossRef]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corra, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef]
- Cardenas, C.; Muller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgo, J.; Diehl, J.A.; et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 2016, 14, 2313–2324. [Google Scholar] [CrossRef]
- Koval, O.M.; Nguyen, E.K.; Santhana, V.; Fidler, T.P.; Sebag, S.C.; Rasmussen, T.P.; Mittauer, D.J.; Strack, S.; Goswami, P.C.; Abel, E.D.; et al. Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.D.; Wu, Y.; Domann, F.E.; Spitz, D.R.; Anderson, M.E. Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS ONE 2014, 9, e96866. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, X.; Shen, Q.; Yang, X.; Yu, C.; Cai, C.; Cai, G.; Meng, X.; Zou, F. Mitochondrial Ca(2)(+) uniporter is critical for store-operated Ca(2)(+) entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 2015, 458, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Bastian, A.; Thorpe, J.E.; Disch, B.C.; Bailey-Downs, L.C.; Gangjee, A.; Devambatla, R.K.; Henthorn, J.; Humphries, K.M.; Vadvalkar, S.S.; Ihnat, M.A. A small molecule with anticancer and antimetastatic activities induces rapid mitochondrial-associated necrosis in breast cancer. J. Pharm. Exp. 2015, 353, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Chen, X.; Cui, W.; Wen, W.; Lu, F.; Sun, X.; Ma, D.; Yuan, Y.; Li, Z.; Hou, N.; et al. RIPK1 Binds MCU to Mediate Induction of Mitochondrial Ca(2+) Uptake and Promotes Colorectal Oncogenesis. Cancer Res. 2018, 78, 2876–2885. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca(2+) levels and tumor growth. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Q.; Zhou, D.; Song, W.; Yang, Q.; Ju, B.; Zhang, L.; Xie, H.; Zhou, L.; Hu, Z.; et al. HINT2 triggers mitochondrial Ca(2+) influx by regulating the mitochondrial Ca(2+) uniporter (MCU) complex and enhances gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett. 2017, 411, 106–116. [Google Scholar] [CrossRef]
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1149–1163. [Google Scholar] [CrossRef]
- Cui, C.; Yang, J.; Fu, L.; Wang, M.; Wang, X. Progress in understanding mitochondrial calcium uniporter complex-mediated calcium signalling: A potential target for cancer treatment. Br. J. Pharmacol. 2019, 176, 1190–1205. [Google Scholar] [CrossRef]
- Nathan, S.R.; Pino, N.W.; Arduino, D.M.; Perocchi, F.; MacMillan, S.N.; Wilson, J.J. Synthetic Methods for the Preparation of a Functional Analogue of Ru360, a Potent Inhibitor of Mitochondrial Calcium Uptake. Inorg. Chem. 2017, 56, 3123–3126. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.X.; Armache, J.P.; Lee, C.; Yang, Y.; Zeng, W.; Mootha, V.K.; Cheng, Y.; Bai, X.C.; Jiang, Y. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature 2018, 559, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Wu, M.; Yin, Y.; Herzik, M.A., Jr.; Lander, G.C.; Lee, S.Y. Cryo-EM structure of a mitochondrial calcium uniporter. Science (New York N.Y.) 2018, 361, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, R.; Wang, C.; Siliciano, A.F.; Long, S.B. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature 2018, 559, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Checchetto, V.; Teardo, E.; Carraretto, L.; Leanza, L.; Szabo, I. Physiology of intracellular potassium channels: A unifying role as mediators of counterion fluxes? Biochim. Biophys. Acta 2016, 1857, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, A.; Bednarczyk, P.; Jedraszko, J.; Kampa, R.P.; Koprowski, P.; Krajewska, M.; Kucman, S.; Kulawiak, B.; Laskowski, M.; Rotko, D.; et al. Mitochondrial potassium channels—An overview. Postepy Biochem. 2018, 64, 196–212. [Google Scholar] [CrossRef]
- Pardo, L.A.; del Camino, D.; Sanchez, A.; Alves, F.; Bruggemann, A.; Beckh, S.; Stuhmer, W. Oncogenic potential of EAG K(+) channels. EMBO J. 1999, 18, 5540–5547. [Google Scholar] [CrossRef]
- Pardo, L.A.; Stuhmer, W. The roles of K(+) channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef]
- Peruzzo, R.; Biasutto, L.; Szabo, I.; Leanza, L. Impact of intracellular ion channels on cancer development and progression. Eur. Biophys. J. 2016. [Google Scholar] [CrossRef]
- Mu, D.; Chen, L.; Zhang, X.; See, L.H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.; Servoss, A.; et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 2003, 3, 297–302. [Google Scholar] [CrossRef]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef] [PubMed]
- Bielanska, J.; Hernandez-Losa, J.; Perez-Verdaguer, M.; Moline, T.; Somoza, R.; Ramon, Y.C.S.; Condom, E.; Ferreres, J.C.; Felipe, A. Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr. Cancer Drug Targets 2009, 9, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; O’Reilly, P.; Doyle, A.; Venturini, E.; Zoratti, M.; Szegezdi, E.; Szabo, I. Correlation between potassium channel expression and sensitivity to drug-induced cell death in tumor cell lines. Curr. Pharm. Des. 2014, 20, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Krabbendam, I.E.; Honrath, B.; Culmsee, C.; Dolga, A.M. Mitochondrial Ca(2+)-activated K(+) channels and their role in cell life and death pathways. Cell Calcium 2018, 69, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Werth, F.; Nguyen, H.A.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef] [PubMed]
- Sassi, N.; De Marchi, U.; Fioretti, B.; Biasutto, L.; Gulbins, E.; Franciolini, F.; Szabo, I.; Zoratti, M. An investigation of the occurrence and properties of the mitochondrial intermediate-conductance Ca2+-activated K+ channel mtKCa3.1. Biochim. Biophys. Acta 2010, 1797, 1260–1267. [Google Scholar] [CrossRef]
- Kovalenko, I.; Glasauer, A.; Schockel, L.; Sauter, D.R.; Ehrmann, A.; Sohler, F.; Hagebarth, A.; Novak, I.; Christian, S. Identification of KCa3.1 Channel as a Novel Regulator of Oxidative Phosphorylation in a Subset of Pancreatic Carcinoma Cell Lines. PLoS ONE 2016, 11, e0160658. [Google Scholar] [CrossRef]
- Szabo, I.; Bock, J.; Grassme, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnino, A.; Manago, A.; Leanza, L.; Gontarewitz, A.; Linder, B.; Azzolini, M.; Biasutto, L.; Zoratti, M.; Peruzzo, R.; Legler, K.; et al. Tumor-reducing effect of the clinically used drug clofazimine in a SCID mouse model of pancreatic ductal adenocarcinoma. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Manago, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531.e10. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Trentin, L.; Becker, K.A.; Frezzato, F.; Zoratti, M.; Semenzato, G.; Gulbins, E.; Szabo, I. Clofazimine, Psora-4 and PAP-1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia 2013, 27, 1782–1785. [Google Scholar] [CrossRef]
- Vennekamp, J.; Wulff, H.; Beeton, C.; Calabresi, P.A.; Grissmer, S.; Hansel, W.; Chandy, K.G. Kv1.3-blocking 5-phenylalkoxypsoralens: A new class of immunomodulators. Mol. Pharmacol. 2004, 65, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.I.; Trapani, V. Multidrug resistance phenotypes and MRS2 mitochondrial magnesium channel: Two players from one stemness? Cancer Biol. 2009, 8, 615–617. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Wei, X.; Yan, P.; Han, Y.; Sun, S.; Wu, K.; Fan, D. Human mitochondrial Mrs2 protein promotes multidrug resistance in gastric cancer cells by regulating p27, cyclin D1 expression and cytochrome C release. Cancer Biol. 2009, 8, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Derdak, Z.; Robson, S.C. Mitochondrial recoupling: A novel therapeutic strategy for cancer? Br. J. Cancer 2011, 105, 469–474. [Google Scholar] [CrossRef]
- Derdak, Z.; Mark, N.M.; Beldi, G.; Robson, S.C.; Wands, J.R.; Baffy, G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008, 68, 2813–2819. [Google Scholar] [CrossRef]
- Yu, G.; Liu, J.; Xu, K.; Dong, J. Uncoupling protein 2 mediates resistance to gemcitabine-induced apoptosis in hepatocellular carcinoma cell lines. Biosci. Rep. 2015, 35, e00231. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Fiorini, C.; Dando, I.; Menegazzi, M.; Sgarbossa, A.; Costanzo, C.; Palmieri, M.; Donadelli, M. Role of mitochondrial uncoupling protein 2 in cancer cell resistance to gemcitabine. Biochim. Biophys. Acta 2012, 1823, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Pons, D.G.; Nadal-Serrano, M.; Torrens-Mas, M.; Valle, A.; Oliver, J.; Roca, P. UCP2 inhibition sensitizes breast cancer cells to therapeutic agents by increasing oxidative stress. Free Radic. Biol. Med. 2015, 86, 67–77. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion Channel/Pore | Channel Localization Within Mitochondria | Drug Affecting Channel/Pore Activity | Effect on Cell Death/ Chemo-Resistance | References |
|---|---|---|---|---|
| BCL-2 anti-apoptotic protein | MOM | Venetoclax (ABT-199) | Kills cancer cells by blocking anti-apoptotic activity of BCL-2 | [43] |
| VDAC1 | MOM | cyathane-type diterpenoid | Kills cancer cells even in the absence of BAX/BAK | [68] |
| VDAC1 | MOM | VDAC1-based peptides | Detaches hexokinase II and BCL-XL/BCL-2 from VDAC and potentiates the effect of chemotherapeutics | [76,77] |
| α7 nAChR | MOM | PNU-282987 | Decreases cytochrome c release stimulated by oxidative stress | [88] |
| α7 nAChR | MOM | nicotine | Confers resistance to cell death induced by gemcitabine | [89] |
| MPTP | IMM | Hirsutine, betulinic acid, honokiol | Activates PTP and counteracts BCL-2/BCL-XL-mediated apoptosis resistance | [112] [113] [114] [115] |
| MCU | IMM | Ru265 | Inhibits MCU—prevents hypoxia-induced injury (not tested on tumor cells) | [142] |
| MCU | IMM | AG311 | Reduces metastasis | [135] |
| IKCa | IMM | TRAM-34 | Sensitizes melanoma cells to vemurafenib | [157] |
| mtKv1.3 | IMM | PAPTP, PCARBTP | Kills various cancer cells independently of p53 mutation and BAX/BCL-2 expression | [163] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peruzzo, R.; Szabo, I. Contribution of Mitochondrial Ion Channels to Chemo-Resistance in Cancer Cells. Cancers 2019, 11, 761. https://doi.org/10.3390/cancers11060761
Peruzzo R, Szabo I. Contribution of Mitochondrial Ion Channels to Chemo-Resistance in Cancer Cells. Cancers. 2019; 11(6):761. https://doi.org/10.3390/cancers11060761
Chicago/Turabian StylePeruzzo, Roberta, and Ildiko Szabo. 2019. "Contribution of Mitochondrial Ion Channels to Chemo-Resistance in Cancer Cells" Cancers 11, no. 6: 761. https://doi.org/10.3390/cancers11060761
APA StylePeruzzo, R., & Szabo, I. (2019). Contribution of Mitochondrial Ion Channels to Chemo-Resistance in Cancer Cells. Cancers, 11(6), 761. https://doi.org/10.3390/cancers11060761