Functional Interaction of Hypoxia-Inducible Factor 2-Alpha and Autophagy Mediates Drug Resistance in Colon Cancer Cells

 , ,
, ,  ,
,  and
and 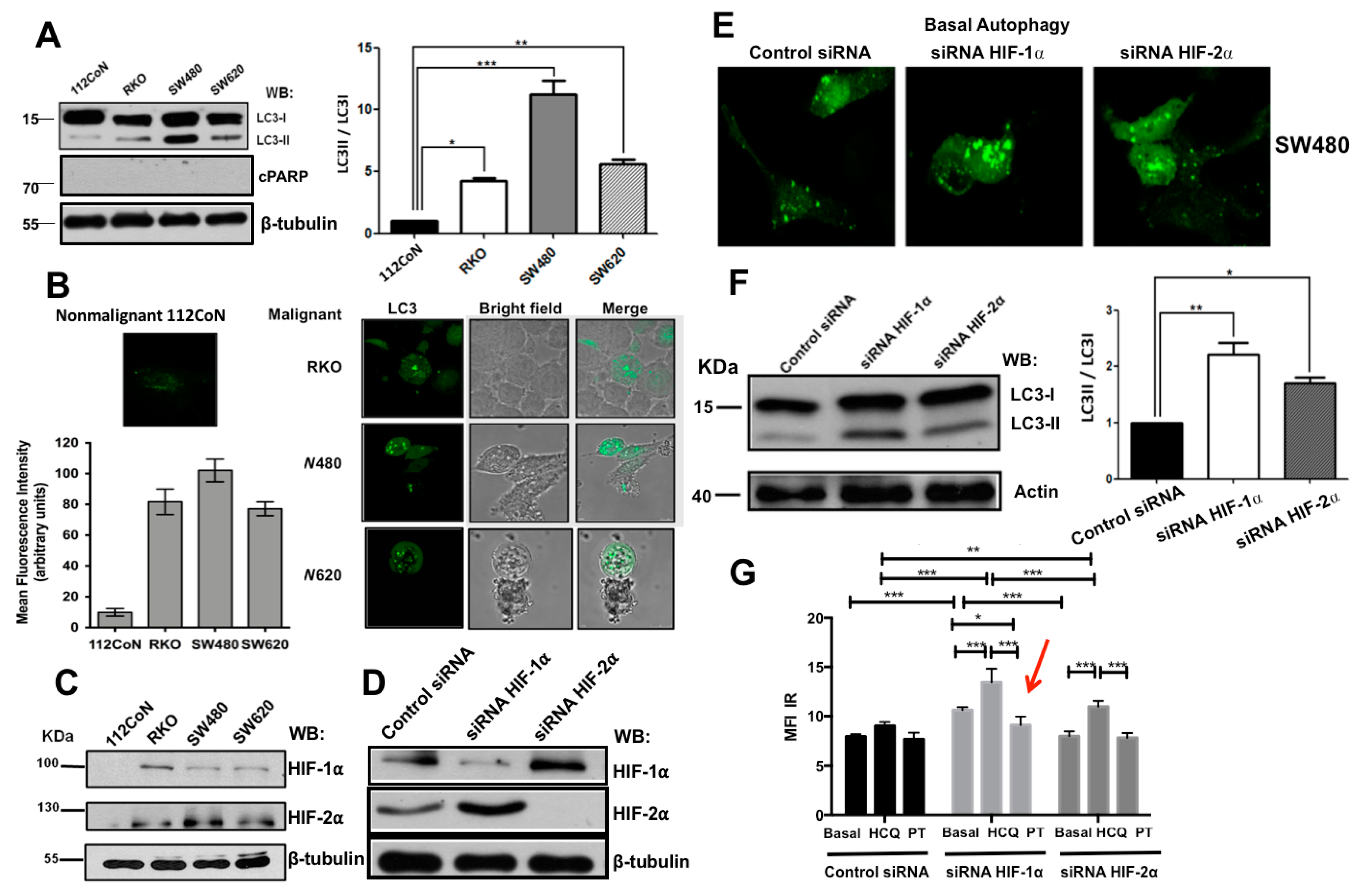{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Human Colon Malignant Cells Show High Levels of Basal Autophagy and Knockdown of Hypoxia-Inducible Factors (HIFs) Expression Increases These Levels under Normoxic Conditions
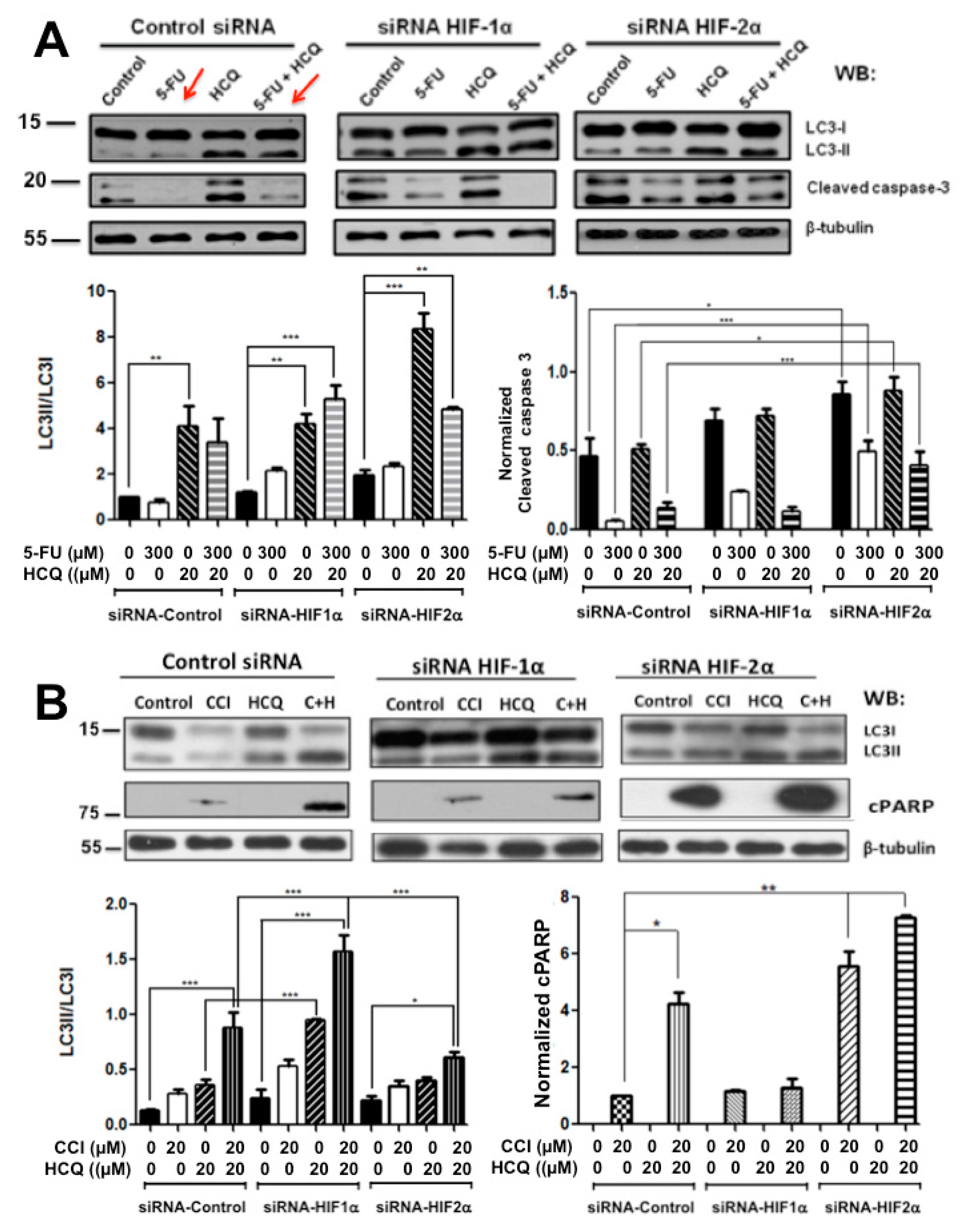
2.2. Colon Cancer Cells Can Be Sensitized to Drug Treatment with Starvation but Combining These Conditions with HIF-2α Silencing Induced Severe Effects on Cell Death
2.3. Resistance to Drug Treatment Can also Be Overcome by Only Combining Treatment with HIF-2α Knockdown Expression
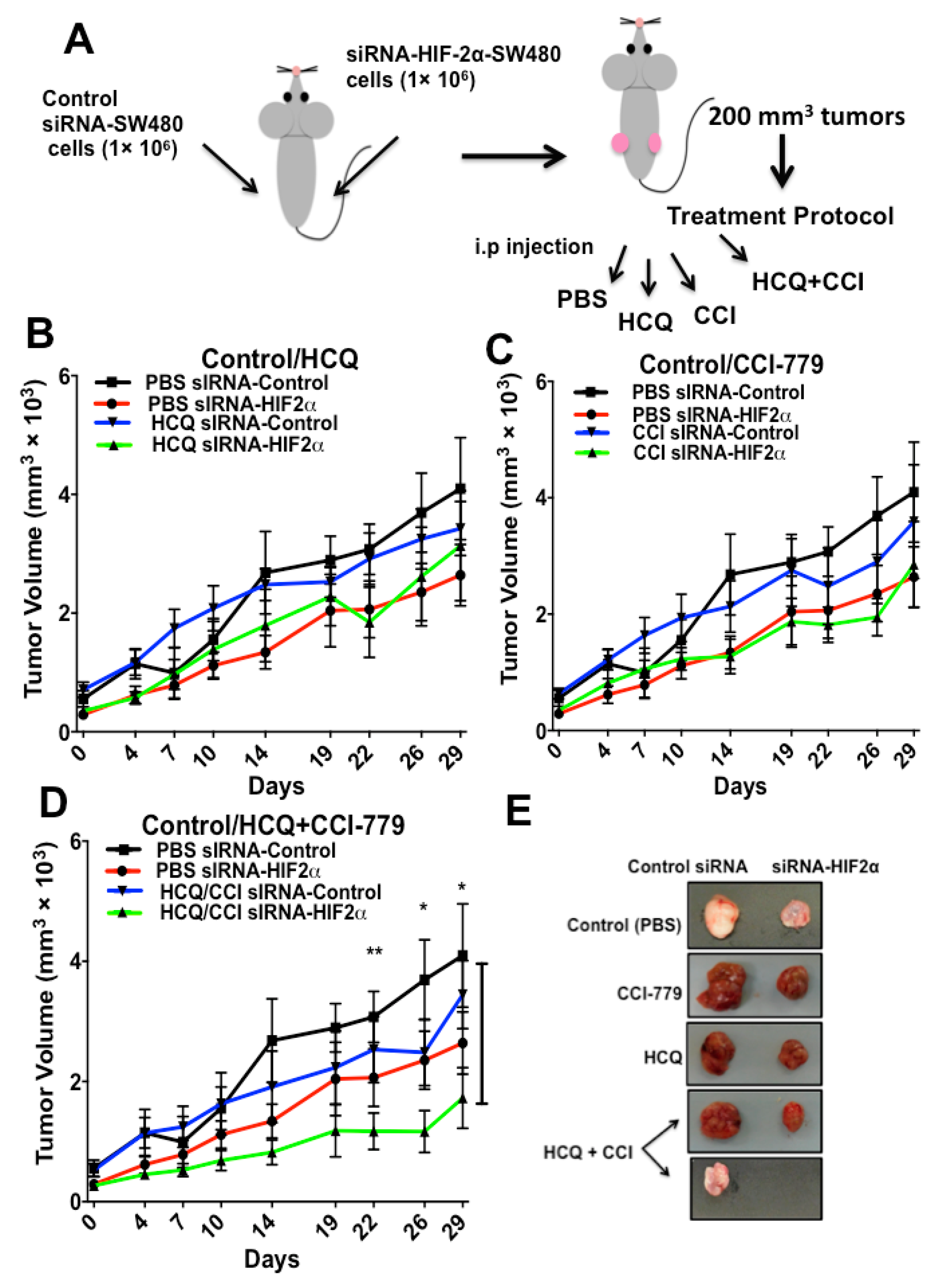
2.4. HIF-2α Blockade Expression Sensitizes Tumors to Treatment with Autophagy and mTOR Inhibitors In Vivo
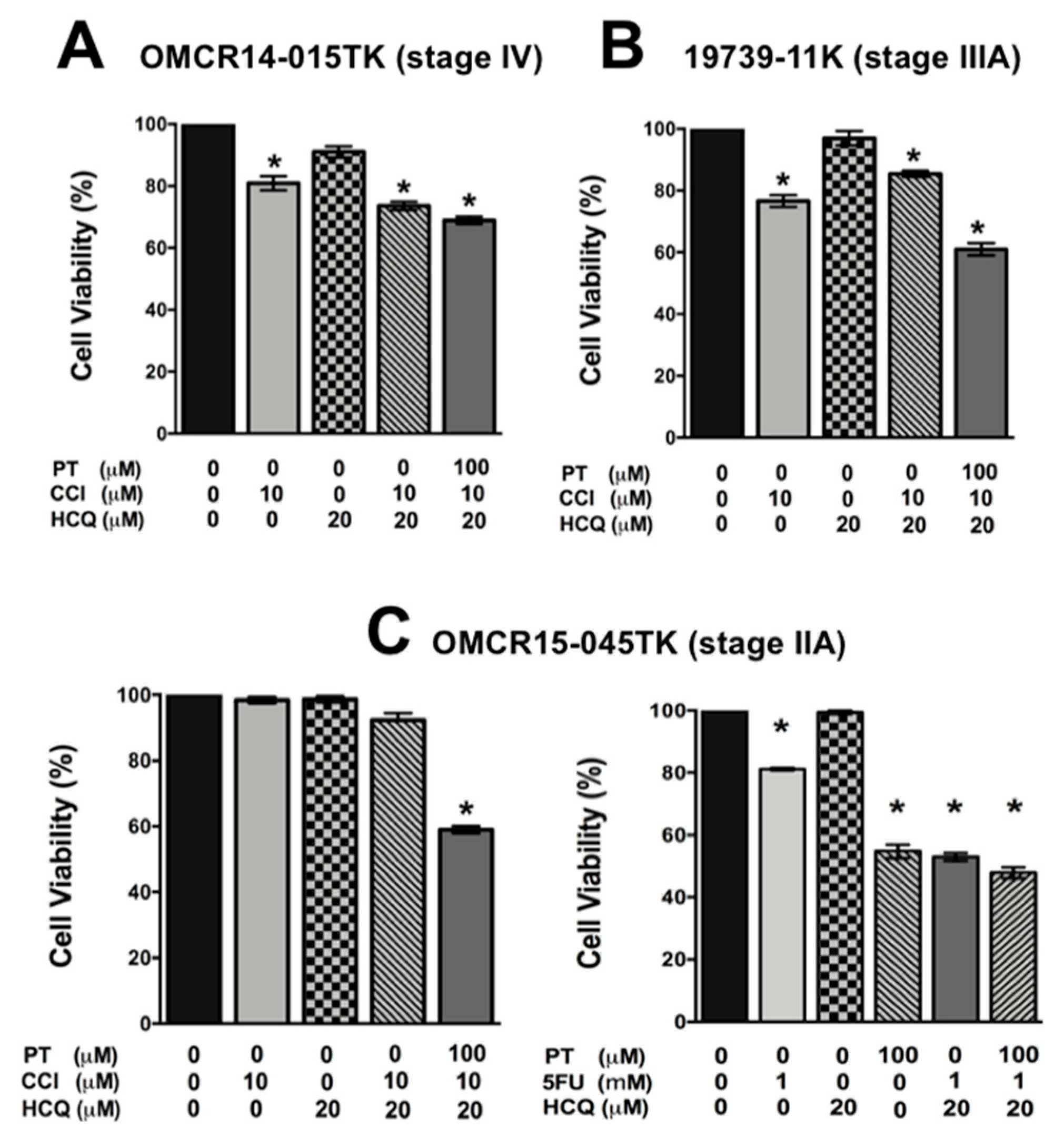
2.5. A Specific HIF-2α Heterodimerization Antagonist also Overcame the Cytotoxic Resistance Displayed by Colon Cancer Patient-Derived Primary Culture Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Ethics Statement
4.3. Plasmids
4.4. Cell Lines
4.5. Primary Cell Cultures Derived from Colorectal Cancer (CRC) Patients’ Tissue Specimens
4.6. Western Blotting and Apoptosis Analysis
4.7. Autophagy Detection
4.8. HIF-1α or HIF-2α Knockdown
4.9. Flow cytometry/ Fluorescence Activated Cell Sorting (FACS) Analysis
4.10. Viability Assay
4.11. Immunofluorescence Analysis
4.12. Xenograft Tumor M Model
4.13. Analysis of Oxidative Stress
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updates 2011, 14, 191–201. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1a and HIF2a: Sibling rivalry in hypoxic tumor growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef]
- Santoyo-Ramos, P.; Likhatcheva, M.; Castañeda-Patlán, C.; García-Zepeda, E.; Robles-Flores, M. Hypoxia-inducible factors participate in the modulation of stemness and malignancy of colon cancer cells playing opposite roles in canonical Wnt signaling. PLoS ONE 2014, 9, e112580. [Google Scholar] [CrossRef]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.M.; Cowen, R.L.; Debray, C.; Eustace, A.; Erler, J.T.; Sheppard, F.C.; Parker, C.A.; Stratford, I.J.; Williams, K.J. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol. Pharmacol. 2006, 69, 411–418. [Google Scholar] [CrossRef]
- Nardinocchi, L.; Puca, R.; Sacchi, A.; D’Orazi, G. Inhibition of HIF-1alpha activity by homeodomain-interacting protein kinase-2 correlates with sensitization of chemoresistant cells to undergo apoptosis. Mol. Cancer 2009, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-L.; Moding, E.J.; Kirsch, D.G. Reining in radiation injury: HIF2α in the gut. Sci. Transl. Med. 2014, 6, 236fs20. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.W.; Su, Y.; Zhu, H.; Cao, J.; Ding, W.J.; Zhao, Y.C.; He, Q.J.; Yang, B. HIF-1 alpha-dependent autophagy protects HeLa cells from fenretinide (4-HPR)-induced apoptosis in hypoxia. Pharmacol. Res. 2010, 62, 416–425. [Google Scholar] [CrossRef]
- Xie, X.; White, E.P.; Mehnert, J.M. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS ONE 2013, 8, e55096. [Google Scholar] [CrossRef] [PubMed]
- Menrad, H.; Werno, C.; Schmid, T.; Copanaki, C.; Deller, Y.; Dhene, N.; Brüne, B. Roles of Hypoxia-Inducible Factor-1α (HIF-1α) versus HIF-2α in the survival of hepatocellular tumor spheroids. Hepatology 2010, 51, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Terkhorn, S.P.; Freeman, T.; Adams, C.S.; Garcia, J.; Shapiero, I.M.; Srinivas, V. Regulation of autophagy in cartilage: HIF-2 suppresses chondrocyte autophagy. Arthritis Rheum. 2009, 60, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.H.; Li, Q.; Ma, H.W.; Key, J.; Zhang, L.; Chen, R.; Garcia, J.A.; Naidoo, J.; Longgood, J.; Frantz, D.E.; et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 2013, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Kubisch, J.; Türei, D.; Földvári-Nagy, L.; Dunai, Z.; Zsákai, L.; Varga, M.; Vellai, T.; Csermely, P.; Korcsmáros, T. Complex regulation of autophagy in cancer –integrated approaches to discover the networks that hold a double-edged sword. Semin. Cancer Biol. 2013, 23, 252–261. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhang, X.; Wang, X.; Sun, B. Autophagy enhances the agressiveness of human colorectal cancer cells and their ability to adapt to apoptotic stimulus. Cancer Biol. Med. 2012, 9, 105–110. [Google Scholar]
- Pietrocola, F.; Izzo, V.; Niso-Santano, M.; Vacchelli, E.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Regulaltion of autophagy by stress-responsive transcription factors. Semin. Cancer Biol. 2013, 23, 310–322. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef]
- Bracht, K.; Nicholls, A.M.; Liu, Y.; Bodmer, W.F. 5-Fluorouracil response in a large panel of colorectal cancer cell lines is associated with mismatch repair deficiency. Br. J. Cancer 2010, 103, 340–346. [Google Scholar] [CrossRef]
- Scortegagna, M.; Ding, K.; Oktay, Y.; Gaur, A.; Thurmond, F.; Yan, L.J.; Marck, B.T.; Matsumoto, A.M.; Shelton, J.M.; Richardson, J.A.; et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat. Genet. 2003, 35, 331–340. [Google Scholar] [CrossRef]
- Bertout, J.A.; Majmundara, A.J.; Gordana, J.D.; Lama, J.C.; Ditswortha, D.; Keith, B.; Browna, E.J.; Nathanson, K.L.; Simon, M.C. HIF2α inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc. Natl. Acad. Sci. USA 2009, 106, 14391–14396. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Yoon, J.S.; Won, Y.; Park, B.; Lee, Y. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. Acta Pathol. Microbiol. Immunol. Scand. 2012, 120, 597–604. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer efffect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Schonewolf, C.A.; Mehta, M.; Schiff, D.; Wu, H.; Haffty, B.G.; Karantza, V.; Jabbour, S.K. Autophagy inhibition by chloroquine sensitizes HT-29 colorectal cancer cells to concurrent chemoradiation. World J. Gastrointest. Oncol. 2014, 6, 74–82. [Google Scholar] [CrossRef]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-Target Efficacy of a HIF2α Antagonist in Preclinical Kidney Cancer Models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.M.; Rizzi, J.P.; Han, G.; When, P.M.; Cao, Z.; Du, X.; Cheng, T.; Czerwinski, R.M.; Dixon, D.D.; Goggin, B.S.; et al. A Small-Molecule Antagonist of HIF2a Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016, 76, 5491–5500. [Google Scholar] [CrossRef]
- Chen, W.; Hill1, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting Renal Cell Carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Saint-Martin, A.; Castañeda-Patlán, M.C.; Robles-Flores, M. The role of hypoxia inducible factors in cancer resistance. J. Cell Signal. 2017, 2, 154. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Heberle, A.M.; Prentzell, M.T.; Van Eunen, K.; Bakker, B.M.; Grellscheld, S.N.; Thedieck, K. Molecular mechanisms of mTOR regulation by stress. Mol. Cell. Oncol. 2015, 2, e970489. [Google Scholar] [CrossRef]
- Carroll, V.A.; Ashcroft, M. Role of hipoxia-inducible factor HIF-1α versus HIF-2α in the regulation of HIF target genes in response to hipoxia, insulin-like growth factor-1, or loss of von Hippel-Lindau function: Implications for targeting the HIF pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef]
- Schulz, K.; Milke, L.; Rübsamen, D.; Menrad, H.; Schmid, Y.; Brüne, B. HIF-1α protein is upregulated in HIF-2α depleted cells via enhanced translation. FEBS Lett. 2012, 586, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Doi, N.; Ogawa, R.; Cui, Z.G.; Morii, A.; Watanabe, A.; Kanayama, S.; Yoneda, Y.; Kondo, T. The acquired radioresistance in HeLa cells under conditions mimicking hypoxia was attenuated by a decreased expression of HIF subunit genes induced by RNA interference. Exp. Cell Res. 2015, 33, 349–360. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Miao, Y.R.; Diep, A.N.; Wu, C.; Rankin, E.B.; Atwood, T.F.; Xing, L.; Giaccia, A.J. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci. Transl. Med. 2014, 6, 236ra64. [Google Scholar] [CrossRef]
- Franovic, A.; Holterman, C.E.; Payette, J.; Lee, S. Human cancers converge at the HIF-2 oncogenic axis. Proc. Natl. Acad. Sci. USA 2009, 106, 21306–21311. [Google Scholar] [CrossRef]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.-J.; Diehl, J.A.; Simon, M.C. HIF-2_ promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, K.; Gao, D.; Zhu, G.; Wu, D.; Wang, X.; Chen, Y.; Du, Y.; Song, W.; Ma, Z.; et al. Reciprocal regulation of hipoxia-inducible factor 2α and GLI1 expression associated with the radioresistance of renal cell carcinoma. Int. J. Radiat. Oncol. 2014, 90, 942–951. [Google Scholar] [CrossRef]
- Koh, M.Y.; Lemos, R., Jr.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1a– to HIF-2a–dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef]
- Yun, Z.; Lin, Q. Hypoxia and regulation of cancer cell stemness. Adv. Exp. Med. Biol. 2014, 772, 41–53. [Google Scholar]
- Imamura, T.; Kikuchi, H.; Herraiz, M.T.; Park, D.Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1α and HIF-2α have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef]
- Munster, T.; Gibbs, J.P.; Shen, D.; Baethge, B.A.; Botstein, G.R.; Caldwell, J.; Dietz, F.; Ettlinger, R.; Golden, H.E.; Lindsley, H.; et al. Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis Rheum. 2002, 46, 1460–1469. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saint-Martin, A.; Martínez-Ríos, J.; Castañeda-Patlán, M.C.; Sarabia-Sánchez, M.A.; Tejeda-Muñoz, N.; Chinney-Herrera, A.; Soldevila, G.; Benelli, R.; Santoyo-Ramos, P.; Poggi, A.; et al. Functional Interaction of Hypoxia-Inducible Factor 2-Alpha and Autophagy Mediates Drug Resistance in Colon Cancer Cells. Cancers 2019, 11, 755. https://doi.org/10.3390/cancers11060755
Saint-Martin A, Martínez-Ríos J, Castañeda-Patlán MC, Sarabia-Sánchez MA, Tejeda-Muñoz N, Chinney-Herrera A, Soldevila G, Benelli R, Santoyo-Ramos P, Poggi A, et al. Functional Interaction of Hypoxia-Inducible Factor 2-Alpha and Autophagy Mediates Drug Resistance in Colon Cancer Cells. Cancers. 2019; 11(6):755. https://doi.org/10.3390/cancers11060755
Chicago/Turabian StyleSaint-Martin, Abril, Jacobo Martínez-Ríos, M. Cristina Castañeda-Patlán, Miguel Angel Sarabia-Sánchez, Nydia Tejeda-Muñoz, Alberto Chinney-Herrera, Gloria Soldevila, Roberto Benelli, Paula Santoyo-Ramos, Alessandro Poggi, and et al. 2019. "Functional Interaction of Hypoxia-Inducible Factor 2-Alpha and Autophagy Mediates Drug Resistance in Colon Cancer Cells" Cancers 11, no. 6: 755. https://doi.org/10.3390/cancers11060755
APA StyleSaint-Martin, A., Martínez-Ríos, J., Castañeda-Patlán, M. C., Sarabia-Sánchez, M. A., Tejeda-Muñoz, N., Chinney-Herrera, A., Soldevila, G., Benelli, R., Santoyo-Ramos, P., Poggi, A., & Robles-Flores, M. (2019). Functional Interaction of Hypoxia-Inducible Factor 2-Alpha and Autophagy Mediates Drug Resistance in Colon Cancer Cells. Cancers, 11(6), 755. https://doi.org/10.3390/cancers11060755