BARD1 is a Low/Moderate Breast Cancer Risk Gene: Evidence Based on an Association Study of the Central European p.Q564X Recurrent Mutation

 , ,
, , 

Abstract
1. Introduction
2. Results
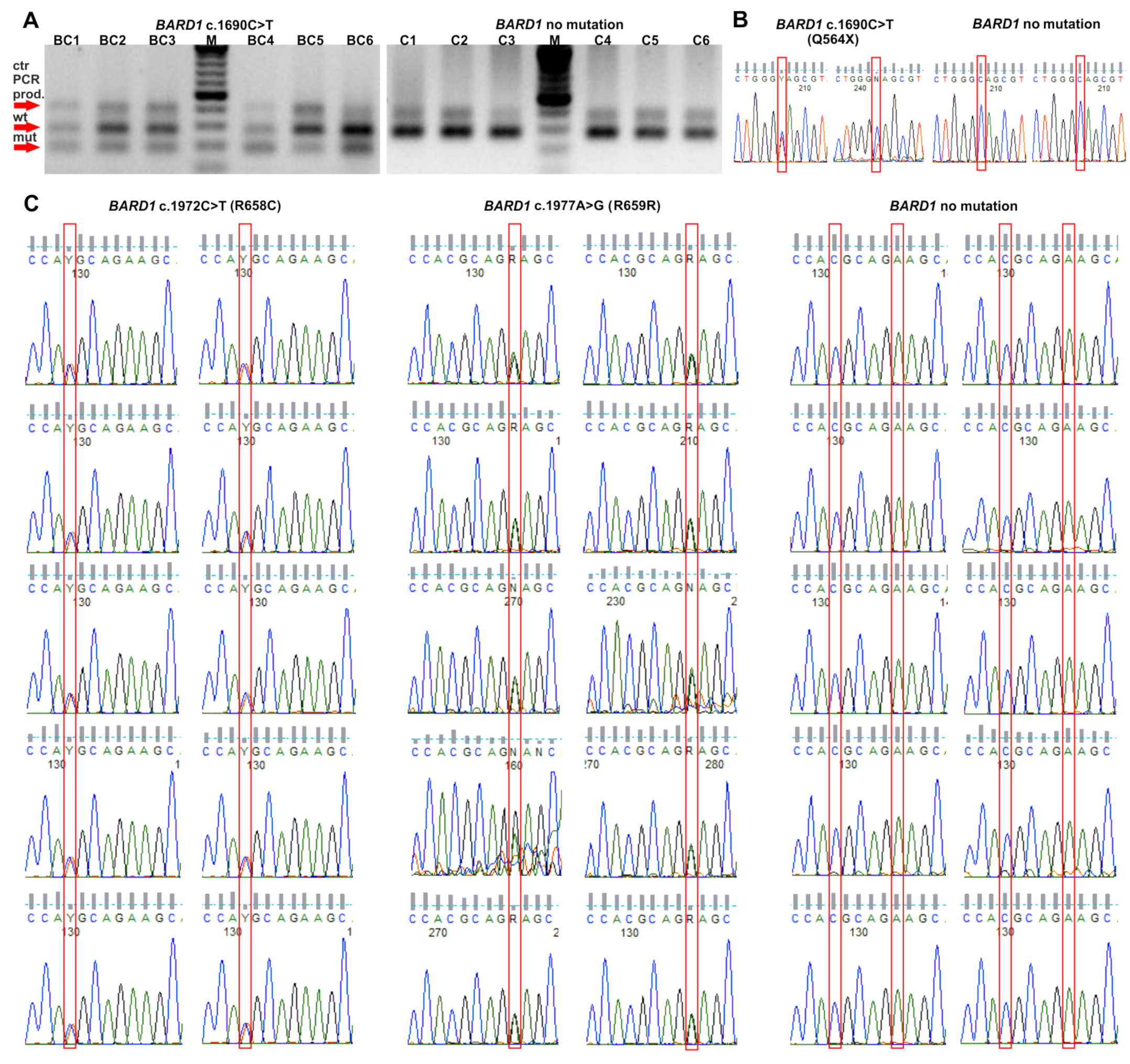
2.1. Selection of BARD1 Variants
2.2. The Frequency of BARD1 Variants and BC Risk Estimates
2.3. Predictive Value of BARD1 Variants
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Variant Screening
4.3. Validation of Genotyping Results
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Guilford, P.; Caldas, C.; International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Fletcher, O.; Palles, C.; Rudd, M.; Webb, E.; Sellick, G.; dos Santos Silva, I.; McCormack, V.; Gibson, L.; Fraser, A.; et al. Counting potentially functional variants in BRCA1, BRCA2 and ATM predicts breast cancer susceptibility. Hum. Mol. Genet. 2007, 16, 1051–1057. [Google Scholar] [CrossRef]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef]
- Kumaran, M.; Krishnan, P.; Cass, C.E.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Breast cancer associated germline structural variants harboring small noncoding RNAs impact post-transcriptional gene regulation. Sci. Rep. 2018, 8, 7529. [Google Scholar] [CrossRef] [PubMed]
- Krepischi, A.C.; Achatz, M.I.; Santos, E.M.; Costa, S.S.; Lisboa, B.C.; Brentani, H.; Santos, T.M.; Goncalves, A.; Nobrega, A.F.; Pearson, P.L.; et al. Germline DNA copy number variation in familial and early-onset breast cancer. Breast Cancer Res. 2012, 14, R24. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium; Craddock, N.; Hurles, M.E.; Cardin, N.; Pearson, R.D.; Plagnol, V.; Robson, S.; Vukcevic, D.; Barnes, C.; Conrad, D.F.; et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3000 shared controls. Nature 2010, 464, 713–720. [Google Scholar] [CrossRef]
- Joo, J.E.; Dowty, J.G.; Milne, R.L.; Wong, E.M.; Dugue, P.A.; English, D.; Hopper, J.L.; Goldgar, D.E.; Giles, G.G.; Southey, M.C.; et al. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat. Commun. 2018, 9, 867. [Google Scholar] [CrossRef]
- Severi, G.; Southey, M.C.; English, D.R.; Jung, C.H.; Lonie, A.; McLean, C.; Tsimiklis, H.; Hopper, J.L.; Giles, G.G.; Baglietto, L. Epigenome-wide methylation in DNA from peripheral blood as a marker of risk for breast cancer. Breast Cancer Res. Treat. 2014, 148, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Ratajska, M.; Pilyugin, M. New concepts on BARD1: Regulator of BRCA pathways and beyond. Int. J. Biochem. Cell. Biol. 2016, 72, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Formicola, D.; Capasso, M. Dualistic Role of BARD1 in Cancer. Genes 2017, 8, 375. [Google Scholar] [CrossRef]
- Feki, A.; Jefford, C.E.; Berardi, P.; Wu, J.Y.; Cartier, L.; Krause, K.H.; Irminger-Finger, I. BARD1 induces apoptosis by catalysing phosphorylation of p53 by DNA-damage response kinase. Oncogene 2005, 24, 3726–3736. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Leung, W.C.; Li, J.; Dubois-Dauphin, M.; Harb, J.; Feki, A.; Jefford, C.E.; Soriano, J.V.; Jaconi, M.; Montesano, R.; et al. Identification of BARD1 as mediator between proapoptotic stress and p53-dependent apoptosis. Mol. Cell. 2001, 8, 1255–1266. [Google Scholar] [CrossRef]
- Thai, T.H.; Du, F.; Tsan, J.T.; Jin, Y.; Phung, A.; Spillman, M.A.; Massa, H.F.; Muller, C.Y.; Ashfaq, R.; Mathis, J.M.; et al. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum. Mol. Genet. 1998, 7, 195–202. [Google Scholar] [CrossRef]
- Ghimenti, C.; Sensi, E.; Presciuttini, S.; Brunetti, I.M.; Conte, P.; Bevilacqua, G.; Caligo, M.A. Germline mutations of the BRCA1-associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosomes Cancer 2002, 33, 235–242. [Google Scholar] [CrossRef]
- Ishitobi, M.; Miyoshi, Y.; Hasegawa, S.; Egawa, C.; Tamaki, Y.; Monden, M.; Noguchi, S. Mutational analysis of BARD1 in familial breast cancer patients in Japan. Cancer Lett. 2003, 200, 1–7. [Google Scholar] [CrossRef]
- Karppinen, S.M.; Heikkinen, K.; Rapakko, K.; Winqvist, R. Mutation screening of the BARD1 gene: Evidence for involvement of the Cys557Ser allele in hereditary susceptibility to breast cancer. J. Med. Genet. 2004, 41, e114. [Google Scholar] [CrossRef]
- Sauer, M.K.; Andrulis, I.L. Identification and characterization of missense alterations in the BRCA1 associated RING domain (BARD1) gene in breast and ovarian cancer. J. Med. Genet. 2005, 42, 633–638. [Google Scholar] [CrossRef]
- Vahteristo, P.; Syrjakoski, K.; Heikkinen, T.; Eerola, H.; Aittomaki, K.; von Smitten, K.; Holli, K.; Blomqvist, C.; Kallioniemi, O.P.; Nevanlinna, H. BARD1 variants Cys557Ser and Val507Met in breast cancer predisposition. Eur. J. Hum. Genet. 2006, 14, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Karppinen, S.M.; Barkardottir, R.B.; Backenhorn, K.; Sydenham, T.; Syrjakoski, K.; Schleutker, J.; Ikonen, T.; Pylkas, K.; Rapakko, K.; Erkko, H.; et al. Nordic collaborative study of the BARD1 Cys557Ser allele in 3956 patients with cancer: Enrichment in familial BRCA1/BRCA2 mutation-negative breast cancer but not in other malignancies. J. Med. Genet. 2006, 43, 856–862. [Google Scholar] [CrossRef][Green Version]
- Huo, X.; Hu, Z.; Zhai, X.; Wang, Y.; Wang, S.; Wang, X.; Qin, J.; Chen, W.; Jin, G.; Liu, J.; et al. Common non-synonymous polymorphisms in the BRCA1 Associated RING Domain (BARD1) gene are associated with breast cancer susceptibility: A case-control analysis. Breast Cancer Res. Treat. 2007, 102, 329–337. [Google Scholar] [CrossRef]
- Jakubowska, A.; Cybulski, C.; Szymanska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Debniak, T.; Gorski, B.; Kowalska, E.; Narod, S.A.; et al. BARD1 and breast cancer in Poland. Breast Cancer Res. Treat. 2008, 107, 119–122. [Google Scholar] [CrossRef]
- Gorringe, K.L.; Choong, D.Y.; Visvader, J.E.; Lindeman, G.J.; Campbell, I.G. BARD1 variants are not associated with breast cancer risk in Australian familial breast cancer. Breast Cancer Res. Treat. 2008, 111, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Johnatty, S.E.; Beesley, J.; Chen, X.; Hopper, J.L.; Southey, M.C.; Giles, G.G.; Goldgar, D.E.; Chenevix-Trench, G.; Spurdle, A.B.; Australian Ovarian Cancer Study Group; et al. The BARD1 Cys557Ser polymorphism and breast cancer risk: An Australian case-control and family analysis. Breast Cancer Res. Treat. 2009, 115, 145–150. [Google Scholar] [CrossRef]
- Guenard, F.; Labrie, Y.; Ouellette, G.; Beauparlant, C.J.; Durocher, F.; BRCAs, I. Genetic sequence variations of BRCA1-interacting genes AURKA, BAP1, BARD1 and DHX9 in French Canadian families with high risk of breast cancer. J. Hum. Genet. 2009, 54, 152–161. [Google Scholar] [CrossRef]
- De Brakeleer, S.; De Greve, J.; Loris, R.; Janin, N.; Lissens, W.; Sermijn, E.; Teugels, E. Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum. Mutat. 2010, 31, E1175–E1185. [Google Scholar] [CrossRef]
- Sabatier, R.; Adelaide, J.; Finetti, P.; Ferrari, A.; Huiart, L.; Sobol, H.; Chaffanet, M.; Birnbaum, D.; Bertucci, F. BARD1 homozygous deletion, a possible alternative to BRCA1 mutation in basal breast cancer. Genes Chromosomes Cancer 2010, 49, 1143–1151. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef]
- Ratajska, M.; Antoszewska, E.; Piskorz, A.; Brozek, I.; Borg, A.; Kusmierek, H.; Biernat, W.; Limon, J. Cancer predisposing BARD1 mutations in breast-ovarian cancer families. Breast Cancer Res. Treat. 2012, 131, 89–97. [Google Scholar] [CrossRef]
- Ratajska, M.; Matusiak, M.; Kuzniacka, A.; Wasag, B.; Brozek, I.; Biernat, W.; Koczkowska, M.; Debniak, J.; Sniadecki, M.; Kozlowski, P.; et al. Cancer predisposing BARD1 mutations affect exon skipping and are associated with overexpression of specific BARD1 isoforms. Oncol. Rep. 2015, 34, 2609–2617. [Google Scholar] [CrossRef]
- Rouleau, E.; Jesson, B.; Briaux, A.; Nogues, C.; Chabaud, V.; Demange, L.; Sokolowska, J.; Coulet, F.; Barouk-Simonet, E.; Bignon, Y.J.; et al. Rare germline large rearrangements in the BRCA1/2 genes and eight candidate genes in 472 patients with breast cancer predisposition. Breast Cancer Res. Treat. 2012, 133, 1179–1190. [Google Scholar] [CrossRef]
- Castera, L.; Krieger, S.; Rousselin, A.; Legros, A.; Baumann, J.J.; Bruet, O.; Brault, B.; Fouillet, R.; Goardon, N.; Letac, O.; et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur. J. Hum. Genet. 2014, 22, 1305–1313. [Google Scholar] [CrossRef]
- Cybulski, C.; Lubinski, J.; Wokolorczyk, D.; Kuzniak, W.; Kashyap, A.; Sopik, V.; Huzarski, T.; Gronwald, J.; Byrski, T.; Szwiec, M.; et al. Mutations predisposing to breast cancer in 12 candidate genes in breast cancer patients from Poland. Clin. Genet. 2015, 88, 366–370. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Slavin, T.P.; Maxwell, K.N.; Lilyquist, J.; Vijai, J.; Neuhausen, S.L.; Hart, S.N.; Ravichandran, V.; Thomas, T.; Maria, A.; Villano, D.; et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017, 3, 22. [Google Scholar] [CrossRef]
- Eliade, M.; Skrzypski, J.; Baurand, A.; Jacquot, C.; Bertolone, G.; Loustalot, C.; Coutant, C.; Guy, F.; Fumoleau, P.; Duffourd, Y.; et al. The transfer of multigene panel testing for hereditary breast and ovarian cancer to healthcare: What are the implications for the management of patients and families? Oncotarget 2017, 8, 1957–1971. [Google Scholar] [CrossRef]
- Thompson, E.R.; Rowley, S.M.; Li, N.; McInerny, S.; Devereux, L.; Wong-Brown, M.W.; Trainer, A.H.; Mitchell, G.; Scott, R.J.; James, P.A.; et al. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J. Clin. Oncol. 2016, 34, 1455–1459. [Google Scholar] [CrossRef]
- Minion, L.E.; Dolinsky, J.S.; Chase, D.M.; Dunlop, C.L.; Chao, E.C.; Monk, B.J. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol. Oncol. 2015, 137, 86–92. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Wubbenhorst, B.; D’Andrea, K.; Garman, B.; Long, J.M.; Powers, J.; Rathbun, K.; Stopfer, J.E.; Zhu, J.; Bradbury, A.R.; et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet. Med. 2015, 17, 630–638. [Google Scholar] [CrossRef]
- Pritzlaff, M.; Summerour, P.; McFarland, R.; Li, S.; Reineke, P.; Dolinsky, J.S.; Goldgar, D.E.; Shimelis, H.; Couch, F.J.; Chao, E.C.; et al. Male breast cancer in a multi-gene panel testing cohort: Insights and unexpected results. Breast Cancer Res. Treat. 2017, 161, 575–586. [Google Scholar] [CrossRef]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meeks, H.; Feng, B.J.; Healey, S.; Thorne, H.; Makunin, I.; Ellis, J.; kConFab, I.; Campbell, I.; Southey, M.; et al. Targeted massively parallel sequencing of a panel of putative breast cancer susceptibility genes in a large cohort of multiple-case breast and ovarian cancer families. J. Med. Genet. 2016, 53, 34–42. [Google Scholar] [CrossRef]
- De Brakeleer, S.; De Greve, J.; Desmedt, C.; Joris, S.; Sotiriou, C.; Piccart, M.; Pauwels, I.; Teugels, E. Frequent incidence of BARD1-truncating mutations in germline DNA from triple-negative breast cancer patients. Clin. Genet. 2016, 89, 336–340. [Google Scholar] [CrossRef]
- Gass, J.; Tatro, M.; Blackburn, P.; Hines, S.; Atwal, P.S. BARD1 nonsense variant c.1921C > T in a patient with recurrent breast cancer. Clin. Case Rep. 2017, 5, 104–107. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.F.; Webb, E.L.; Matakidou, A.; Sellick, G.S.; Williams, R.D.; Bridle, H.; Eisen, T.; Houlston, R.S.; Consortium, G. Variants in the GH-IGF axis confer susceptibility to lung cancer. Genome Res. 2006, 16, 693–701. [Google Scholar] [CrossRef]
- Esteban-Jurado, C.; Vila-Casadesus, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Munoz, J.; Ocana, T.; Balaguer, F.; Lopez-Ceron, M.; et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 2015, 17, 131–142. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef]
- Lincoln, S.E.; Kobayashi, Y.; Anderson, M.J.; Yang, S.; Desmond, A.J.; Mills, M.A.; Nilsen, G.B.; Jacobs, K.B.; Monzon, F.A.; Kurian, A.W.; et al. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More Than 1000 Patients. J. Mol. Diagn. 2015, 17, 533–544. [Google Scholar] [CrossRef]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.; Faust, U.; Sturm, M.; Hackmann, K.; Grundmann, K.; Harmuth, F.; Bosse, K.; Kehrer, M.; Benkert, T.; Klink, B.; et al. HBOC multi-gene panel testing: Comparison of two sequencing centers. Breast Cancer Res. Treat. 2015, 152, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.W.; Hare, E.E.; Mills, M.A.; Kingham, K.E.; McPherson, L.; Whittemore, A.S.; McGuire, V.; Ladabaum, U.; Kobayashi, Y.; Lincoln, S.E.; et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J. Clin. Oncol. 2014, 32, 2001–2009. [Google Scholar] [CrossRef]
- Kwong, A.; Shin, V.Y.; Au, C.H.; Law, F.B.; Ho, D.N.; Ip, B.K.; Wong, A.T.; Lau, S.S.; To, R.M.; Choy, G.; et al. Detection of Germline Mutation in Hereditary Breast and/or Ovarian Cancers by Next-Generation Sequencing on a Four-Gene Panel. J. Mol. Diagn. 2016, 18, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Sandler, G.; Sobolev, R.; Kim, S.H.; Chambers, R.; Bassett, R.Y.; Martineau, J.; Sapra, K.J.; Boyd, L.; Curtin, J.P.; et al. Multigene panels in Ashkenazi Jewish patients yield high rates of actionable mutations in multiple non-BRCA cancer-associated genes. Gynecol. Oncol. 2017, 146, 123–128. [Google Scholar] [CrossRef]
- Crawford, B.; Adams, S.B.; Sittler, T.; van den Akker, J.; Chan, S.; Leitner, O.; Ryan, L.; Gil, E.; van ‘t Veer, L. Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res. Treat. 2017, 163, 383–390. [Google Scholar] [CrossRef]
- Frey, M.K.; Kim, S.H.; Bassett, R.Y.; Martineau, J.; Dalton, E.; Chern, J.Y.; Blank, S.V. Rescreening for genetic mutations using multi-gene panel testing in patients who previously underwent non-informative genetic screening. Gynecol. Oncol. 2015, 139, 211–215. [Google Scholar] [CrossRef]
- Lhota, F.; Zemankova, P.; Kleiblova, P.; Soukupova, J.; Vocka, M.; Stranecky, V.; Janatova, M.; Hartmannova, H.; Hodanova, K.; Kmoch, S.; et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin. Genet. 2016, 90, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Churpek, J.E.; Marquez, R.; Neistadt, B.; Claussen, K.; Lee, M.K.; Churpek, M.M.; Huo, D.; Weiner, H.; Bannerjee, M.; Godley, L.A.; et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer 2016, 122, 304–311. [Google Scholar] [CrossRef]
- Shirts, B.H.; Casadei, S.; Jacobson, A.L.; Lee, M.K.; Gulsuner, S.; Bennett, R.L.; Miller, M.; Hall, S.A.; Hampel, H.; Hisama, F.M.; et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet. Med. 2016, 18, 974–981. [Google Scholar] [CrossRef]
- Suszynska, M.; Klonowska, K.; Jasinska, A.J.; Kozlowski, P. Large-scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes - Providing evidence of cancer predisposition genes. Gynecol. Oncol. 2019. [Google Scholar] [CrossRef]
- Taylor, A.; Brady, A.F.; Frayling, I.M.; Hanson, H.; Tischkowitz, M.; Turnbull, C.; Side, L.; UK Cancer Genetics Group. Consensus for genes to be included on cancer panel tests offered by UK genetics services: guidelines of the UK Cancer Genetics Group. J. Med. Genet. 2018, 55, 372–377. [Google Scholar] [CrossRef]
- Klonowska, K.; Ratajska, M.; Czubak, K.; Kuzniacka, A.; Brozek, I.; Koczkowska, M.; Sniadecki, M.; Debniak, J.; Wydra, D.; Balut, M.; et al. Analysis of large mutations in BARD1 in patients with breast and/or ovarian cancer: The Polish population as an example. Sci. Rep. 2015, 5, 10424. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Bogdanova, N.; Cybulski, C.; Bermisheva, M.; Datsyuk, I.; Yamini, P.; Hillemanns, P.; Antonenkova, N.N.; Khusnutdinova, E.; Lubinski, J.; Dork, T. A nonsense mutation (E1978X) in the ATM gene is associated with breast cancer. Breast Cancer Res. Treat. 2009, 118, 207–211. [Google Scholar] [CrossRef]
- Cybulski, C.; Carrot-Zhang, J.; Kluzniak, W.; Rivera, B.; Kashyap, A.; Wokolorczyk, D.; Giroux, S.; Nadaf, J.; Hamel, N.; Zhang, S.; et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet. 2015, 47, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Noskowicz, M.; Bogdanova, N.; Bermisheva, M.; Takhirova, Z.; Antonenkova, N.; Khusnutdinova, E.; Bremer, M.; Christiansen, H.; Park-Simon, T.W.; Hillemanns, P.; et al. Prevalence of PALB2 mutation c.509_510delGA in unselected breast cancer patients from Central and Eastern Europe. Fam. Cancer 2014, 13, 137–142. [Google Scholar] [CrossRef]
- Cybulski, C.; Kluzniak, W.; Huzarski, T.; Wokolorczyk, D.; Kashyap, A.; Jakubowska, A.; Szwiec, M.; Byrski, T.; Debniak, T.; Gorski, B.; et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: A prospective cohort analysis. Lancet Oncol. 2015, 16, 638–644. [Google Scholar] [CrossRef]
- Pelttari, L.M.; Kiiski, J.; Nurminen, R.; Kallioniemi, A.; Schleutker, J.; Gylfe, A.; Aaltonen, L.A.; Leminen, A.; Heikkila, P.; Blomqvist, C.; et al. A Finnish founder mutation in RAD51D: Analysis in breast, ovarian, prostate, and colorectal cancer. J. Med. Genet. 2012, 49, 429–432. [Google Scholar] [CrossRef]
- Gayther, S.A.; Warren, W.; Mazoyer, S.; Russell, P.A.; Harrington, P.A.; Chiano, M.; Seal, S.; Hamoudi, R.; van Rensburg, E.J.; Dunning, A.M.; et al. Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nat. Genet. 1995, 11, 428–433. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D.; Breast Cancer Linkage, C. Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am. J. Hum. Genet. 2001, 68, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, J.; Phelan, C.M.; Ghadirian, P.; Lynch, H.T.; Garber, J.; Weber, B.; Tung, N.; Horsman, D.; Isaacs, C.; Monteiro, A.N.; et al. Cancer variation associated with the position of the mutation in the BRCA2 gene. Fam. Cancer 2004, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, N.V.; Antonenkova, N.N.; Rogov, Y.I.; Karstens, J.H.; Hillemanns, P.; Dork, T. High frequency and allele-specific differences of BRCA1 founder mutations in breast cancer and ovarian cancer patients from Belarus. Clin. Genet. 2010, 78, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.B.J.; Weber-Lassalle, K.; Klaschik, K.; Neidhardt, G.; Ernst, C.; Blümcke, B.; Klonowska, K.; Volk, A.; Kubisch, C.; Baber, R.; et al. Germline loss-of-function variants in the BARD1 gene are associated with familial breast cancer. Senologie Zeitschrift für Mammadiagnostik und Therapie 2018, 15, e51. [Google Scholar] [CrossRef]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. J. Natl. Cancer Inst. 2018. [Google Scholar] [CrossRef]
- Castera, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Pharoah, P.D.; McMullan, G.; Day, N.E.; Ponder, B.A.; Easton, D. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet. Epidemiol. 2001, 21, 1–18. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Antoniou, A.; Bobrow, M.; Zimmern, R.L.; Easton, D.F.; Ponder, B.A. Polygenic susceptibility to breast cancer and implications for prevention. Nat. Genet. 2002, 31, 33–36. [Google Scholar] [CrossRef]
- Medrano, R.F.; de Oliveira, C.A. Guidelines for the tetra-primer ARMS-PCR technique development. Mol. Biotechnol. 2014, 56, 599–608. [Google Scholar] [CrossRef]


{kind=link}
| Population | Database (Population) | p.Q564X M+/N (%) | p.R658C M+/N (%) | p.R659R M+/N (%) |
|---|---|---|---|---|
| European | EVS (European American) | 0/4300 (−) | 67/4300 (1.558) | 28/4300 (0.651) |
| FLOSSIES (European) | 1/7325 (0.014) | 130/7325 (1.775) | 75/7325 (1.024) | |
| ExAC (European non-Finnish) | 6/33,368 (0.018) | 535/33,352 (1.604) | 196/33,357 (0.588) | |
| gnomAD (European non-Finnish) | 7/64,526 (0.011) | 1078/64,546 (1.670) | 397/64,564 (0.615) | |
| Other | gnomAD (All populations) | 7/141,343 (0.005) | 2302/141,376 (1.628) | 575/141,400 (0.407) |
| gnomAD (European Finnish) | 0/12,543 (−) | 316/12,557 (2.516) | 33/12,559 (0.263) | |
| gnomAD (Ashkenazi Jewish) | 0/5184 (−) | 0/5183 (−) | 81/5183 (1.563) | |
| gnomAD (East Asian) | 0/9977 (−) | 224/9977 (2.245) | 0/9977 (−) | |
| gnomAD (South Asian) | 0/15,307 (−) | 124/15,306 (0.810) | 1/15,306 (0.007) | |
| gnomAD (African) | 0/12,481 (−) | 59/12,481 (0.473) | 9/12,484 (0.072) | |
| gnomAD (Latino) | 0/17,719 (−) | 444/17,718 (2.506) | 30/17,719 (0.169) | |
| gnomAD (Other) | 0/3606 (−) | 57/3608 (1.580) | 24/3608 (0.665) |
| Study Population | Group | p.Q564X | p.R658C | p.R659R | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M + /N (%) | OR | 95%CI | p-Value | M+/N (%) | OR | 95%CI | p-Value | M+/N (%) | OR | 95%CI | p-Value | ||
| Controls | P | 7/4707 (0.15) | - | - | - | 26/4707 (0.55) | - | - | - | 14/4707 (0.30) | - | - | - |
| B | 0/1189 (0.00) | - | - | - | - | - | - | - | - | - | - | - | |
| P + B | 7/5896 (0.12) | - | - | - | - | - | - | - | - | - | - | - | |
| All BC patients | P | 34/12,476 (0.27) | 1.83 | 0.81–4.14 | 0.14 | 80/12,476 (0.64) | 1.16 | 0.75–1.81 | 0.51 | 49/12,476 (0.39) | 1.32 | 0.73–2.40 | 0.36 |
| B | 4/1459 (0.27) | n.a. | n.a. | n.a. | - | - | - | - | - | - | - | - | |
| P + B | 38/13,935 (0.27) | 2.30 | 1.03–5.15 | 0.04 | - | - | - | - | - | - | - | - | |
| 2.24 * | 0.99–5.03 * | 0.05 * | - | - | - | - | - | - | |||||
| 2.12 ** | 0.97–4.62 ** | 0.06 ** | - | - | - | - | - | - | |||||
| Feature | Group | p.Q564X | p.R658C | p.R659R | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M+/N (%) | OR | 95%CI | p-Value | M+/N (%) | OR | 95%CI | p-Value | M+/N (%) | OR | 95%CI | p-Value | ||
| TNBC | P | 6/1120 (0.54) | 3.62 | 1.21–10.78 | 0.02 | 6/1120 (0.54) | 0.97 | 0.40–2.36 | 0.95 | 2/1120 (0.18) | 0.60 | 0.14–2.64 | 0.50 |
| Bilateral BC | P | 2/447 (0.45) | 3.02 | 0.63–14.57 | 0.17 | 4/447 (0.89) | 1.63 | 0.56–4.68 | 0.37 | 0/447 (0.00) | 0.36 | 0.02–6.07 | 0.48 |
| P + B | 3/498 (0.60) | 5.10 | 1.31–19.78 | 0.02 | - | - | - | - | - | - | - | - | |
| 4.85 * | 1.24–18.93 * | 0.02 * | - | - | - | - | - | - | |||||
| 5.22 ** | 1.50–18.19 ** | 0.01 ** | - | - | - | - | - | - | |||||
| BC diagnosed ≤40 y.o. | P | 4/1286 (0.31) | 2.09 | 0.61–7.17 | 0.24 | 7/1286 (0.54) | 0.99 | 0.43–2.28 | 0.97 | 5/1286 (0.39) | 1.30 | 0.47–3.64 | 0.61 |
| P + B | 5/1640 (0.30) | 2.57 | 0.82–8.12 | 0.11 | - | - | - | - | - | - | - | - | |
| 2.60 * | 0.82–8.21 * | 0.10 * | - | - | - | - | - | - | |||||
| 2.66 ** | 0.88–8.03 ** | 0.08 ** | - | - | - | - | - | - | |||||
| ≥1 BC/OC relatives | P | 5/2191 (0.23) | 1.54 | 0.49–4.84 | 0.46 | 14/2191 (0.64) | 1.16 | 0.60–2.22 | 0.66 | 3/2191 (0.14) | 0.46 | 0.13–1.60 | 0.22 |
| P + B | 6/2620 (0.23) | 1.93 | 0.65–5.75 | 0.24 | - | - | - | - | - | - | - | - | |
| 1.88 * | 0.63–5.60 * | 0.26 * | - | - | - | - | - | - | |||||
| 1.92 ** | 0.67–5.46 ** | 0.22 ** | - | - | - | - | - | - | |||||
| “Hereditary” BC risk patients *** | P | 13/4130 (0.31) | 2.12 | 0.85–5.32 | 0.11 | 30/4130 (0.73) | 1.32 | 0.78–2.23 | 0.31 | 9/4130 (0.22) | 0.73 | 0.32–1.69 | 0.47 |
| P + B | 16/4600 (0.35) | 2.94 | 1.21–7.14 | 0.02 | - | - | - | - | - | - | - | - | |
| 2.93 * | 1.20–7.19 * | 0.02 * | - | - | - | - | - | - | |||||
| 2.77 ** | 1.18–6.49 ** | 0.02 ** | - | - | - | - | - | - | |||||
| Feature | POLISH Group n = 12,476 | BELARUSIAN Group n = 1459 | ||
|---|---|---|---|---|
| n of Cases in Which Feature Status was Determined | n and (%) of Positive Cases | n of Cases in Which Feature Status was Determined | n and (%) of Positive Cases | |
| Age at diagnosis (years) | ||||
| ≤40 | 12,459 | 1286 (10.3) | 1459 | 354 (24.3) |
| 41–50 | 12,459 | 4394 (35.3) | 1459 | 520 (35.6) |
| 51–60 | 12,459 | 3248 (26.1) | 1459 | 329 (22.5) |
| 61–70 | 12,459 | 2226 (17.9) | 1459 | 176 (12.1) |
| ≥71 | 12,459 | 1305 (10.5) | 1459 | 80 (5.5) |
| Number of relatives with BC | ||||
| 0 | 11,564 | 9593 (83.0) | 1459 | 1214 (83.2) |
| 1 | 11,564 | 1508 (13.0) | 1459 | 236 (16.2) |
| 2 | 11,564 | 344 (3.0) | 1459 | 9 (0.6) |
| ≥3 | 11,564 | 119 (1.0) | 1459 | 0 (0.0) |
| Numbers of relatives with OC | ||||
| 0 | 11,564 | 11,263 (97.4) | 1459 | 1457 (99.9) |
| ≥1 | 11,564 | 301 (2.6) | 1459 | 2 (0.1) |
| Histological type of BC | ||||
| Ductal, grade 3 | 8670 | 1998 (23.0) | 525 | 42 (8.0) |
| Ductal, grade 1–2 | 8670 | 4057 (46.8) | 525 | 179 (34.1) |
| Ductal, grade unknown | 8670 | 664 (7.7) | 525 | 165 (31.4) |
| Medullary | 8670 | 287 (3.3) | 525 | 10 (1.9) |
| Lobular | 8670 | 1243 (14.3) | 525 | 108 (20.6) |
| Tubulolobular | 8670 | 116 (1.3) | 525 | 21 (4.0) |
| DCIS with microinvasion | 8670 | 305 (3.5) | 525 | 0 (0.0) |
| Molecular type of BC | ||||
| Oestrogen receptor-positive | 8572 | 5970 (69.6) | 875 | 676 (77.3) |
| Progesterone receptor-positive | 8270 | 5885 (71.2) | n.d. | n.d. |
| HER2-positive | 7215 | 1270 (17.6) | n.d. | n.d. |
| TNBC | 6937 | 1120 (16.1) | n.d. | n.d. |
| Size (cm) | ||||
| <1 | 7909 | 914 (11.6) | n.d. | n.d. |
| 1–1.9 | 7909 | 3204 (40.5) | n.d. | n.d. |
| 2–4.9 | 7909 | 3462 (43.8) | n.d. | n.d. |
| ≥5 | 7909 | 329 (4.2) | n.d. | n.d. |
| Other features | ||||
| Bilateral BC | 9787 | 447 (4.6) | 1459 | 51 (3.5) |
| Lymph node-positive | 8162 | 3551 (43.5) | n.d. | n.d. |
| Vital status (deceased) | 12,349 | 2066 (16.7) | n.d. | n.d. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suszynska, M.; Kluzniak, W.; Wokolorczyk, D.; Jakubowska, A.; Huzarski, T.; Gronwald, J.; Debniak, T.; Szwiec, M.; Ratajska, M.; Klonowska, K.; et al. BARD1 is a Low/Moderate Breast Cancer Risk Gene: Evidence Based on an Association Study of the Central European p.Q564X Recurrent Mutation. Cancers 2019, 11, 740. https://doi.org/10.3390/cancers11060740
Suszynska M, Kluzniak W, Wokolorczyk D, Jakubowska A, Huzarski T, Gronwald J, Debniak T, Szwiec M, Ratajska M, Klonowska K, et al. BARD1 is a Low/Moderate Breast Cancer Risk Gene: Evidence Based on an Association Study of the Central European p.Q564X Recurrent Mutation. Cancers. 2019; 11(6):740. https://doi.org/10.3390/cancers11060740
Chicago/Turabian StyleSuszynska, Malwina, Wojciech Kluzniak, Dominika Wokolorczyk, Anna Jakubowska, Tomasz Huzarski, Jacek Gronwald, Tadeusz Debniak, Marek Szwiec, Magdalena Ratajska, Katarzyna Klonowska, and et al. 2019. "BARD1 is a Low/Moderate Breast Cancer Risk Gene: Evidence Based on an Association Study of the Central European p.Q564X Recurrent Mutation" Cancers 11, no. 6: 740. https://doi.org/10.3390/cancers11060740
APA StyleSuszynska, M., Kluzniak, W., Wokolorczyk, D., Jakubowska, A., Huzarski, T., Gronwald, J., Debniak, T., Szwiec, M., Ratajska, M., Klonowska, K., Narod, S., Bogdanova, N., Dörk, T., Lubinski, J., Cybulski, C., & Kozlowski, P. (2019). BARD1 is a Low/Moderate Breast Cancer Risk Gene: Evidence Based on an Association Study of the Central European p.Q564X Recurrent Mutation. Cancers, 11(6), 740. https://doi.org/10.3390/cancers11060740