ROR1 Potentiates FGFR Signaling in Basal-Like Breast Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
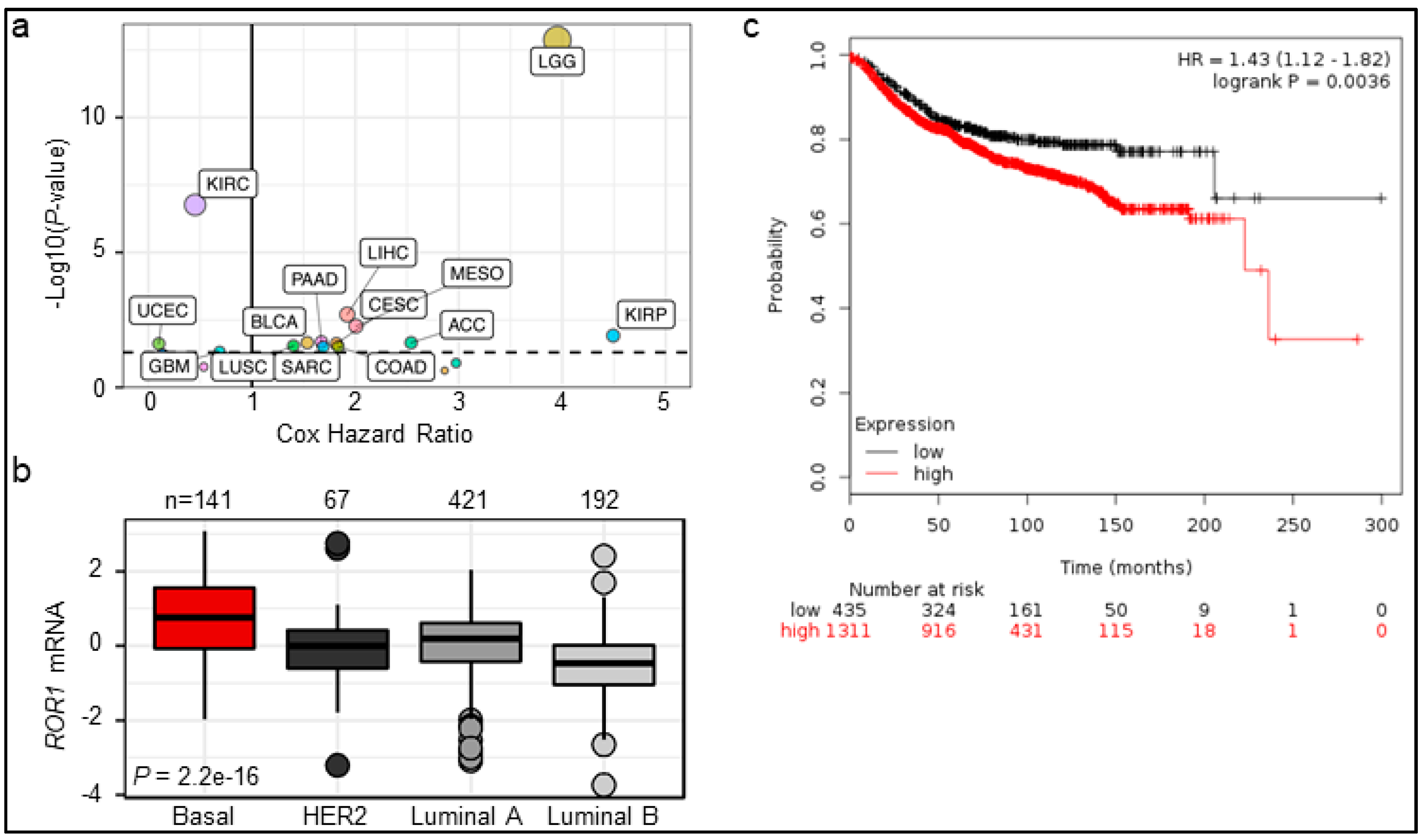
2.1. ROR1 Expression Is Correlated with Poor Overall Survival in Certain Cancers
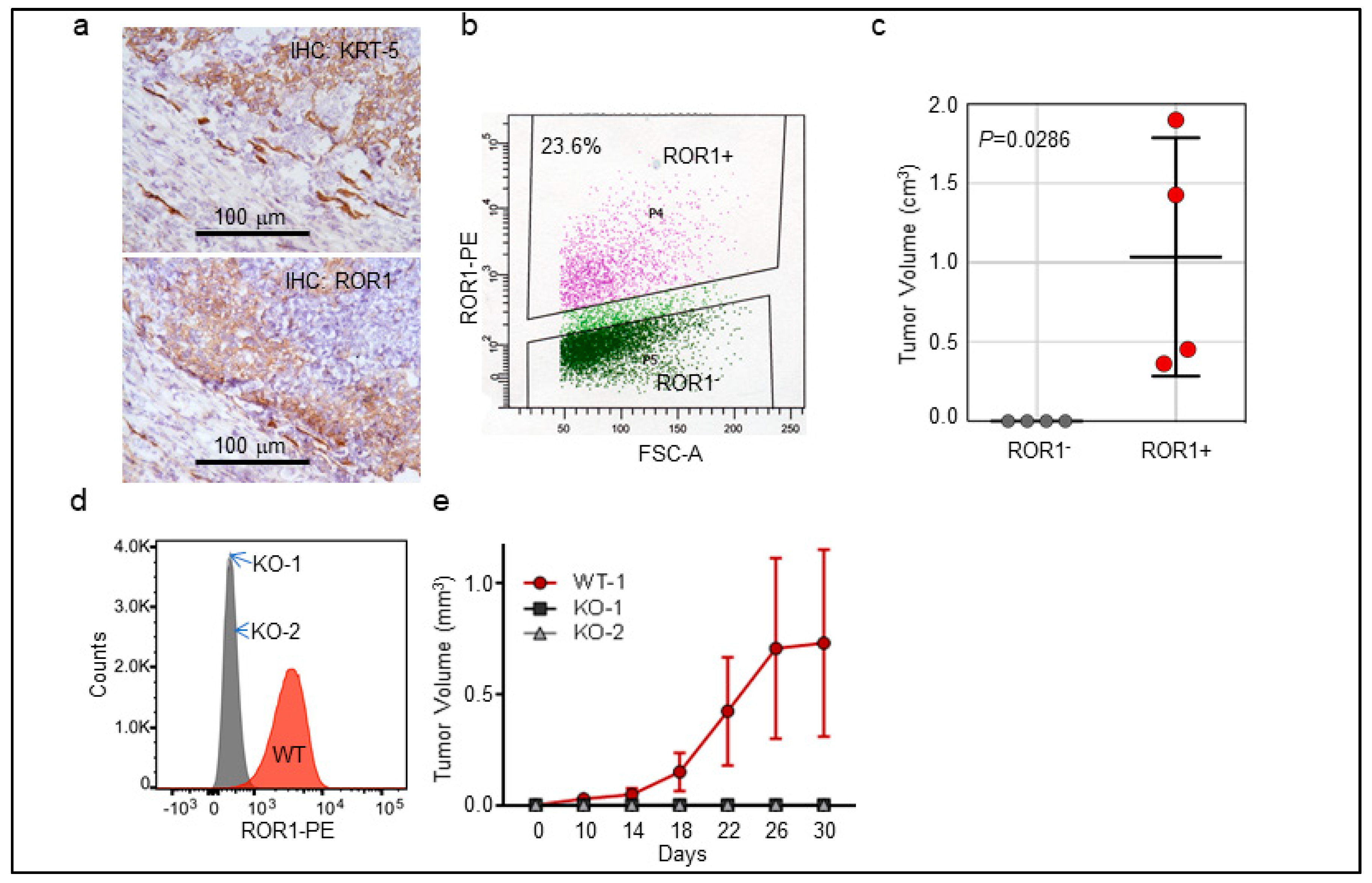
2.2. ROR1+ Cells Are Tumorigenic
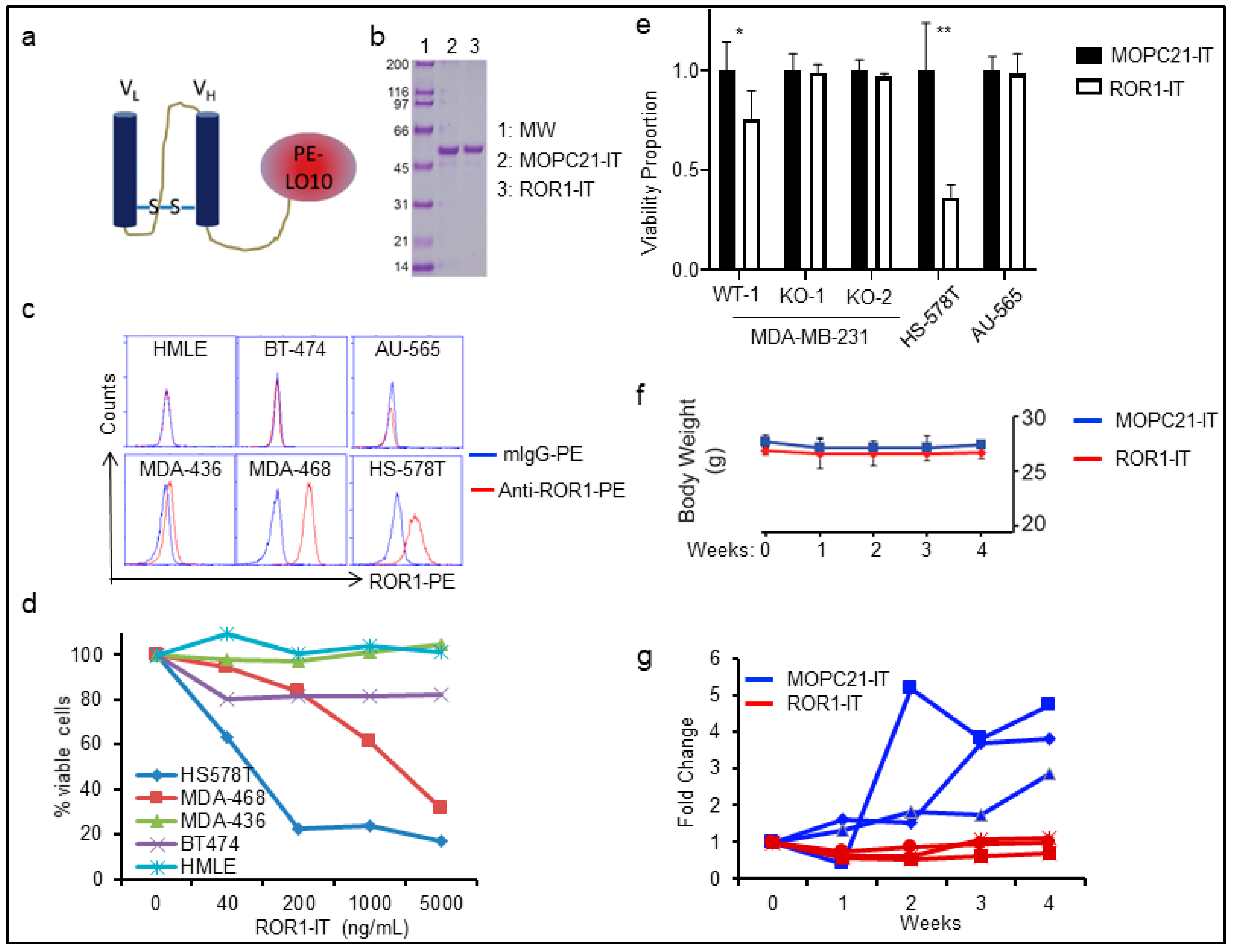
2.3. Targeting ROR1+ Cells Using Immunotoxin in BLBC Therapy
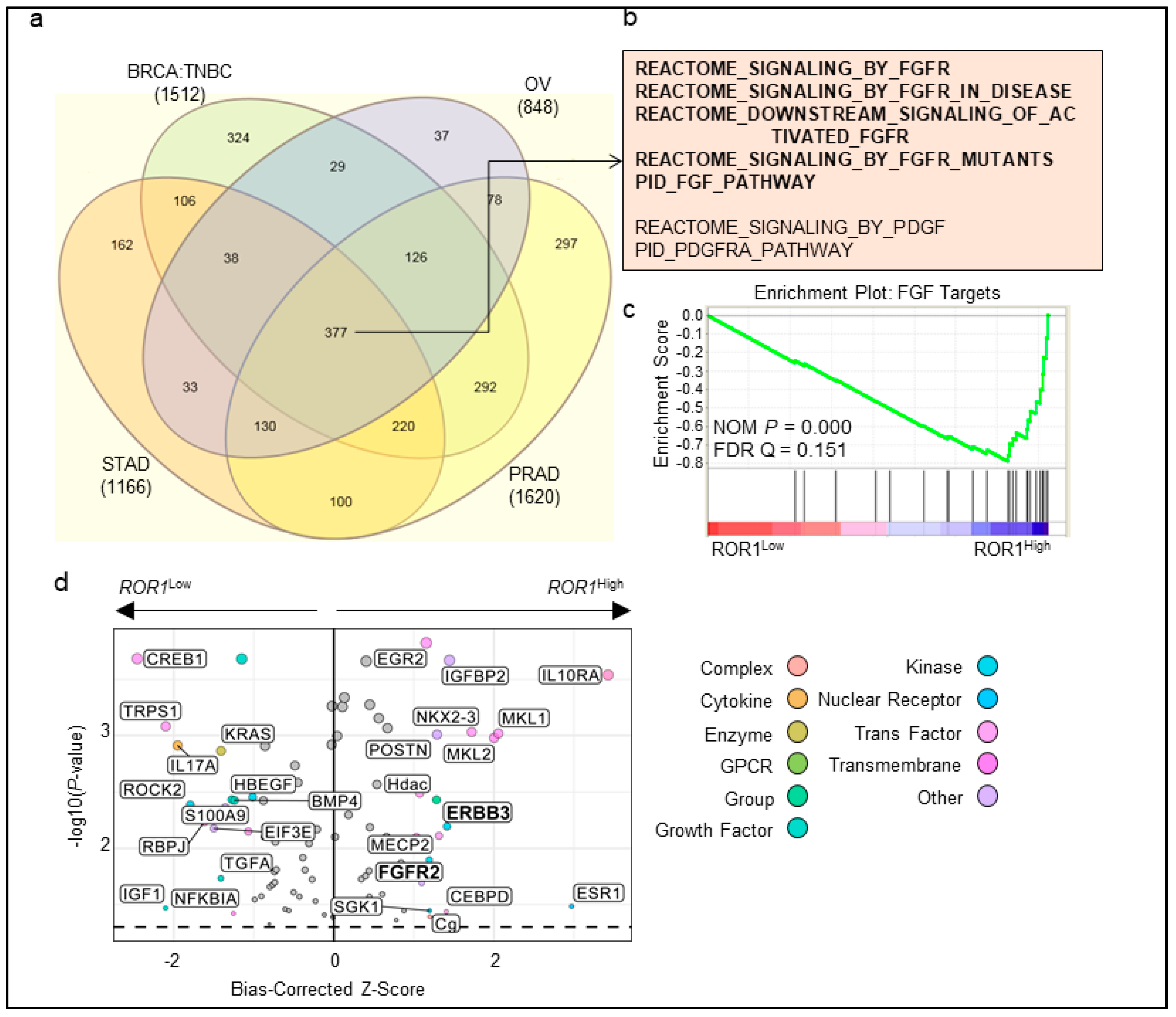
2.4. ROR1 Is Associated with FGFR Activation in Several Major Cancer Types
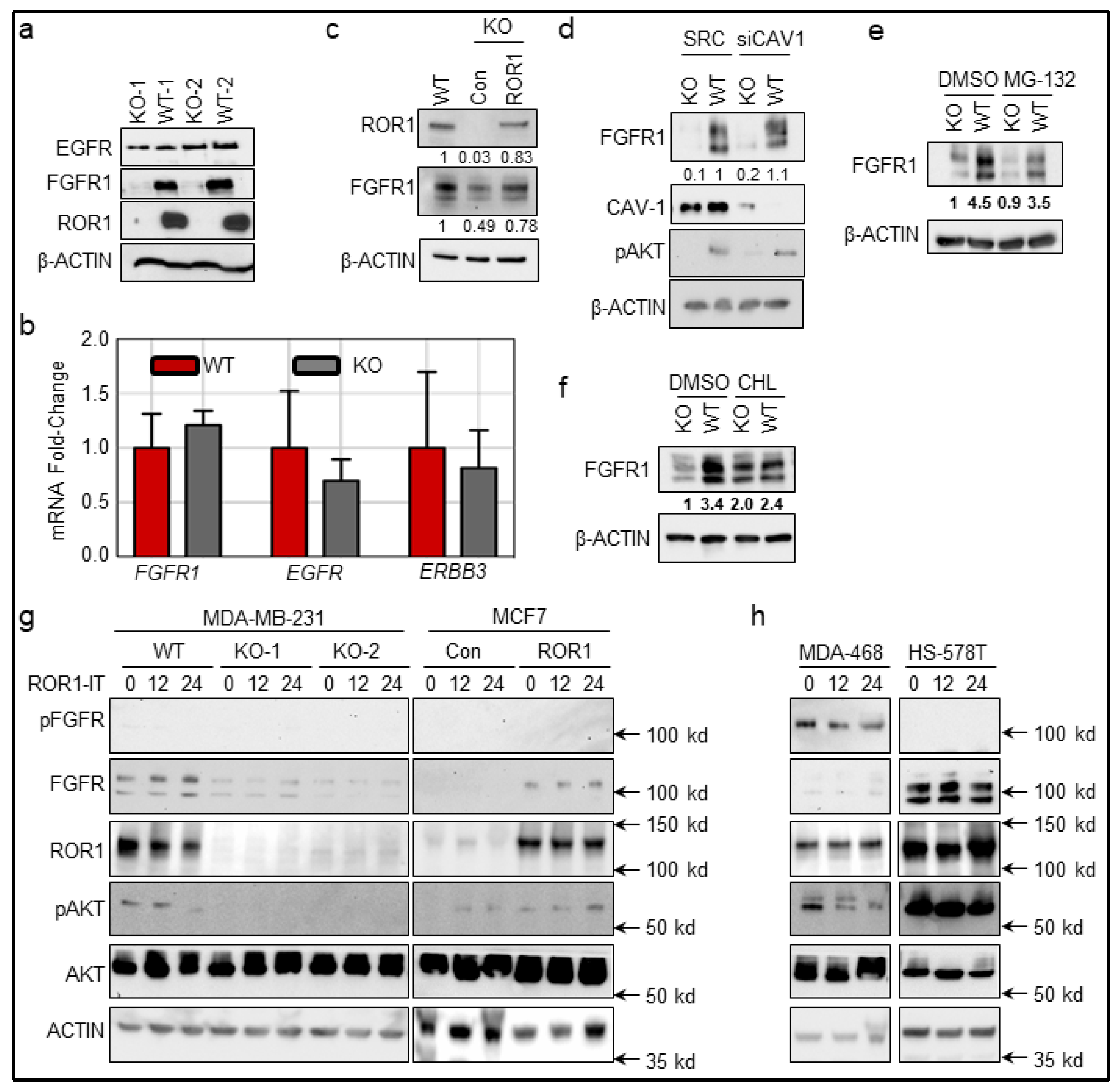
2.5. ROR1 Stabilizes FGFR at the Post-Translational Level
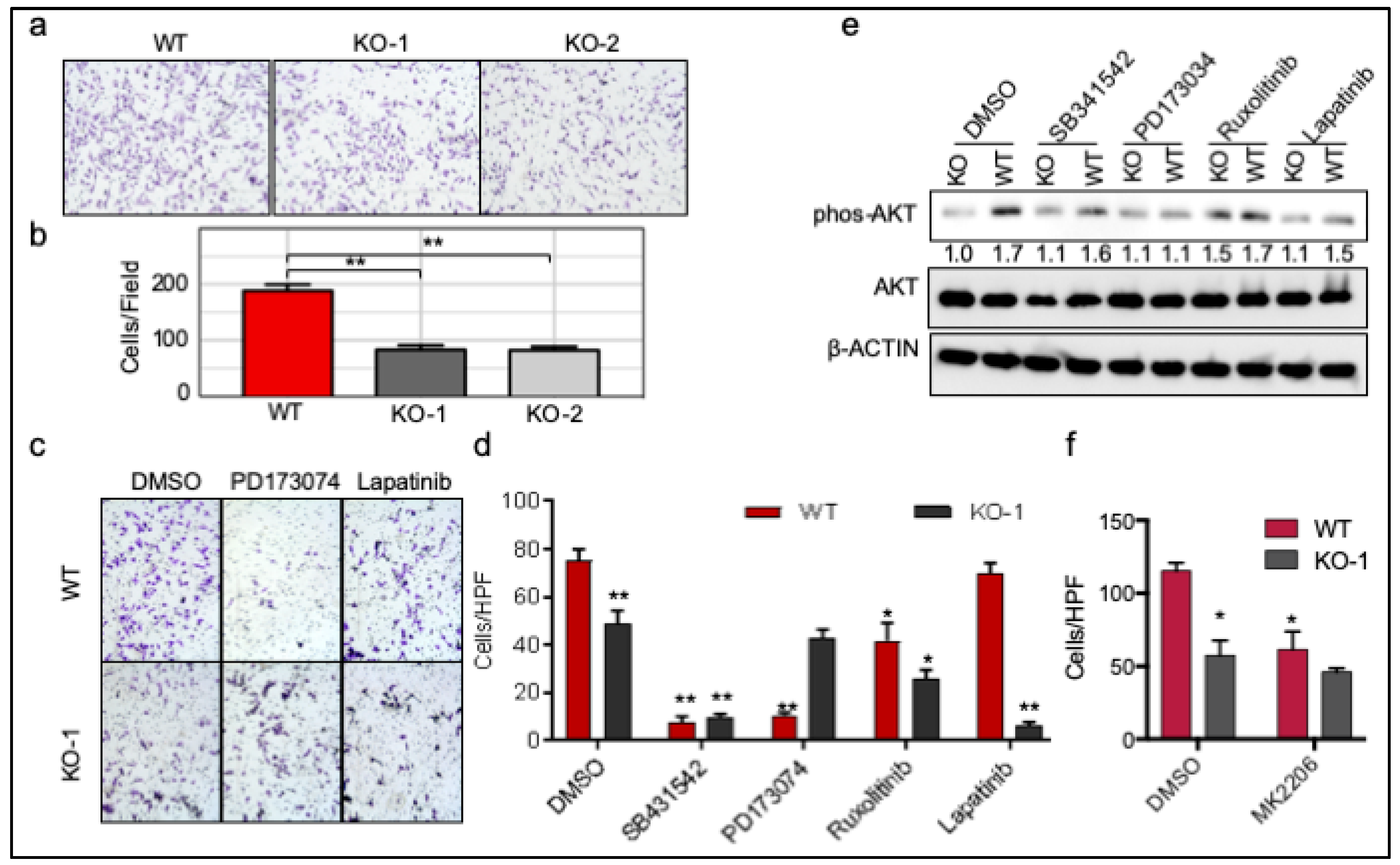
2.6. ROR1 Promotes Invasion through FGFR1
3. Discussion
4. Materials and Methods
4.1. TCGA Analysis
4.2. Pathway Analysis
4.3. Cell Culture and Transfection
4.4. Construction and Purification of Immunotoxin
4.5. In Vitro Invasion Assay
4.6. Flow Cytometry
4.7. CRISPR/Cas9
4.8. Western Blotting
4.9. Real Time PCR
4.10. Immunohistochemistry
4.11. Patient-Derived Xenografts Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Rakha, E.A.; Ellis, I.O. Triple-negative/basal-like breast cancer: review. Pathology 2009, 41, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Brown, J.; Dent, R.; Jackisch, C.; Mackey, J.; Pivot, X.; Steger, G.G.; Suter, T.M.; Toi, M.; Parmar, M.; et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, phase 3 trial. Lancet Oncol. 2013, 14, 933–942. [Google Scholar] [CrossRef]
- Miller, K.; O’Neill, A.M.; Dang, C.T.; Northfelt, D.W.; Gradishar, W.J.; Goldstein, L.J.; Mayer, I.A.; Brufsky, A.; Bloom, S.H.; Sparano, J.A.; et al. Bevacizumab (Bv) in the adjuvant treatment of HER2-negative breast cancer: Final results from Eastern Cooperative Oncology Group E5103. J. Clin. Oncol. 2014, 32, 500. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef]
- Borcherding, N.; Kusner, D.; Liu, G.H.; Zhang, W. ROR1, an embryonic protein with an emerging role in cancer biology. Protein Cell 2014, 5, 496–502. [Google Scholar] [CrossRef]
- Fukuda, T.; Chen, L.; Endo, T.; Tang, L.; Lu, D.; Castro, J.E.; Widhopf, G.F., 2nd; Rassenti, L.Z.; Cantwell, M.J.; Prussak, C.E.; et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc. Natl. Acad. Sci. USA 2008, 105, 3047–3052. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Goodpaster, T.; Randolph-Habecker, J.; Hoffstrom, B.G.; Jalikis, F.G.; Koch, L.K.; Berger, C.; Kosasih, P.L.; Rajan, A.; Sommermeyer, D.; et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin. Cancer Res. 2017, 23, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, B.; Lai, H.; Liu, G.; Ghia, E.M.; Widhopf, G.F., 2nd; Zhang, Z.; Wu, C.C.; Chen, L.; Wu, R.; et al. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 17266–17271. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, L.; Wang-Rodriguez, J.; Zhang, L.; Cui, B.; Frankel, W.; Wu, R.; Kipps, T.J. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am. J. Pathol. 2012, 181, 1903–1910. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, L.; Cui, B.; Chuang, H.Y.; Yu, J.; Wang-Rodriguez, J.; Tang, L.; Chen, G.; Basak, G.W.; Kipps, T.J. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS ONE 2012, 7, e31127. [Google Scholar] [CrossRef] [PubMed]
- Broome, H.E.; Rassenti, L.Z.; Wang, H.Y.; Meyer, L.M.; Kipps, T.J. ROR1 is expressed on hematogones (non-neoplastic human B-lymphocyte precursors) and a minority of precursor-B acute lymphoblastic leukemia. Leuk. Res. 2011, 35, 1390–1394. [Google Scholar] [CrossRef]
- Cui, B.; Zhang, S.; Chen, L.; Yu, J.; Widhopf, G.F., 2nd; Fecteau, J.F.; Rassenti, L.Z.; Kipps, T.J. Targeting ROR1 inhibits epithelial-mesenchymal transition and metastasis. Cancer Res. 2013, 73, 3649–3660. [Google Scholar] [CrossRef]
- Li, C.; Wang, S.; Xing, Z.; Lin, A.; Liang, K.; Song, J.; Hu, Q.; Yao, J.; Chen, Z.; Park, P.K.; et al. A ROR1-HER3-lncRNA signalling axis modulates the Hippo-YAP pathway to regulate bone metastasis. Nat. Cell Biol. 2017, 19, 106–119. [Google Scholar] [CrossRef]
- Choi, M.Y.; Widhopf, G.F., 2nd; Ghia, E.M.; Kidwell, R.L.; Hasan, M.K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E.; et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 2018, 22, 951–959 e953. [Google Scholar] [CrossRef]
- Baskar, S.; Wiestner, A.; Wilson, W.H.; Pastan, I.; Rader, C. Targeting malignant B cells with an immunotoxin against ROR1. MAbs 2012, 4, 349–361. [Google Scholar] [CrossRef]
- Rottman, J.B.; Ganley, K.; Daly, B.; Horton, H.M.; Friedman, K.M.; Perkins, M.; Grande, S.; Rhodes, C.J.; Morgan, R.A.; Horvath, C.J. ROR1-Directed Chimeric Antigen Receptor T Cell Recognition of Self-Antigen Is Associated with Acute Toxicity, T Cell Dysfunction, and Poor Tumor Control. Am. Soc. Hematol. 2017, 130 (Suppl. 1), 4450. [Google Scholar]
- Blankenship, J.W.; Misher, L.; Mitchell, D.; Zhang, N.; Tan, P.; Hoyos, G.H.; Ravikumar, P.; Bader, R.; McMahan, C.J.; Miller, R.E.; et al. Abstract 4995: Anti-ROR1 x anti-CD3 ADAPTIR™ molecule, ES425, redirects T-cell cytotoxicity and inhibits tumor growth in preclinical models of triple-negative breast cancer. Cancer Res. 2016, 76, 4995. [Google Scholar] [CrossRef]
- Deniger, D.C.; Yu, J.; Huls, M.H.; Figliola, M.J.; Mi, T.; Maiti, S.N.; Widhopf, G.F., 2nd; Hurton, L.V.; Thokala, R.; Singh, H.; et al. Sleeping Beauty Transposition of Chimeric Antigen Receptors Targeting Receptor Tyrosine Kinase-Like Orphan Receptor-1 (ROR1) into Diverse Memory T-Cell Populations. PLoS ONE 2015, 10, e0128151. [Google Scholar] [CrossRef]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of Targeting ROR1 in Primates with Chimeric Antigen Receptor–Modified T Cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Hojjat-Farsangi, M.; Daneshmanesh, A.H.; Khan, A.S.; Shetye, J.; Mozaffari, F.; Kharaziha, P.; Rathje, L.S.; Kokhaei, P.; Hansson, L.; Vagberg, J.; et al. First-in-class oral small molecule inhibitor of the tyrosine kinase ROR1 (KAN0439834) induced significant apoptosis of chronic lymphocytic leukemia cells. Leukemia 2018. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef]
- Pond, A.C.; Bin, X.; Batts, T.; Roarty, K.; Hilsenbeck, S.; Rosen, J.M. Fibroblast growth factor receptor signaling is essential for normal mammary gland development and stem cell function. Stem Cells 2013, 31, 178–189. [Google Scholar] [CrossRef]
- Brady, N.; Chuntova, P.; Bade, L.K.; Schwertfeger, K.L. The FGF/FGFR axis as a therapeutic target in breast cancer. Expert Rev. Endocrinol. Metab. 2013, 8, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Issa, A.; Gill, J.W.; Heideman, M.R.; Sahin, O.; Wiemann, S.; Dey, J.H.; Hynes, N.E. Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast Cancer Res. 2013, 15, R8. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Lu, C.; Ida, L.; Yanagisawa, K.; Usukura, J.; Cheng, J.; Hotta, N.; Shimada, Y.; Isomura, H.; Suzuki, M.; et al. ROR1 sustains caveolae and survival signalling as a scaffold of cavin-1 and caveolin-1. Nat. Commun. 2016, 7, 10060. [Google Scholar] [CrossRef] [PubMed]
- Lanczky, A.; Nagy, A.; Bottai, G.; Munkacsy, G.; Szabo, A.; Santarpia, L.; Gyorffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar] [CrossRef]
- Weldon, J.E.; Xiang, L.; Chertov, O.; Margulies, I.; Kreitman, R.J.; FitzGerald, D.J.; Pastan, I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009, 113, 3792–3800. [Google Scholar] [CrossRef]
- Hasan, M.K.; Yu, J.; Widhopf, G.F., 2nd; Rassenti, L.Z.; Chen, L.; Shen, Z.; Briggs, S.P.; Neuberg, D.S.; Kipps, T.J. Wnt5a induces ROR1 to recruit DOCK2 to activate Rac1/2 in chronic lymphocytic leukemia. Blood 2018, 132, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, L.; Chen, Y.; Hasan, M.K.; Ghia, E.M.; Zhang, L.; Wu, R.; Rassenti, L.Z.; Widhopf, G.F.; Shen, Z.; et al. Wnt5a induces ROR1 to associate with 14-3-3zeta for enhanced chemotaxis and proliferation of chronic lymphocytic leukemia cells. Leukemia 2017, 31, 2608–2614. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, L.; Cui, B.; Widhopf, G.F., 2nd; Shen, Z.; Wu, R.; Zhang, L.; Zhang, S.; Briggs, S.P.; Kipps, T.J. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J. Clin. Investig. 2016, 126, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Yanagisawa, K.; Sugiyama, R.; Hosono, Y.; Shimada, Y.; Arima, C.; Kato, S.; Tomida, S.; Suzuki, M.; Osada, H.; et al. NKX2-1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 2012, 21, 348–361. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Malecki, J.; Bjorklund, S.M.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Park, S.; Shu, S.; He, L.; Kong, W.; Zhang, W.; Yuan, Z.; Wang, L.H.; Cheng, J.Q. Advances of AKT pathway in human oncogenesis and as a target for anti-cancer drug discovery. Curr. Cancer Drug Targets 2008, 8, 2–6. [Google Scholar] [PubMed]
- Cheng, G.Z.; Zhang, W.; Wang, L.H. Regulation of cancer cell survival, migration, and invasion by Twist: AKT2 comes to interplay. Cancer Res. 2008, 68, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.; Pearson, A.; Herrera-Abreu, M.T.; Johnson, D.; Mackay, A.; Welti, J.C.; Natrajan, R.; Reynolds, A.R.; Reis-Filho, J.S.; Ashworth, A.; et al. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 2011, 17, 5275–5286. [Google Scholar] [CrossRef]
- Perez-Garcia, J.; Munoz-Couselo, E.; Soberino, J.; Racca, F.; Cortes, J. Targeting FGFR pathway in breast cancer. Breast 2018, 37, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Kostas, M.; Haugsten, E.M.; Zhen, Y.; Sorensen, V.; Szybowska, P.; Fiorito, E.; Lorenz, S.; Jones, N.; de Souza, G.A.; Wiedlocha, A.; et al. Protein Tyrosine Phosphatase Receptor Type G (PTPRG) Controls Fibroblast Growth Factor Receptor (FGFR) 1 Activity and Influences Sensitivity to FGFR Kinase Inhibitors. Mol. Cell. Proteomics 2018, 17, 850–870. [Google Scholar] [CrossRef]
- Shirley, M. Olaratumab: First Global Approval. Drugs 2017, 77, 107–112. [Google Scholar] [CrossRef]
- Daneshmanesh, A.H.; Hojjat-Farsangi, M.; Ghaderi, A.; Moshfegh, A.; Hansson, L.; Schultz, J.; Vagberg, J.; Bystrom, S.; Olsson, E.; Olin, T.; et al. A receptor tyrosine kinase ROR1 inhibitor (KAN0439834) induced significant apoptosis of pancreatic cells which was enhanced by erlotinib and ibrutinib. PLoS ONE 2018, 13, e0198038. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, G.; Borcherding, N.; Kolb, R.; Kluz, P.; Li, W.; Sugg, S.; Zhang, J.; Lai, D.A.; Zhang, W. ROR1 Potentiates FGFR Signaling in Basal-Like Breast Cancer. Cancers 2019, 11, 718. https://doi.org/10.3390/cancers11050718
Pandey G, Borcherding N, Kolb R, Kluz P, Li W, Sugg S, Zhang J, Lai DA, Zhang W. ROR1 Potentiates FGFR Signaling in Basal-Like Breast Cancer. Cancers. 2019; 11(5):718. https://doi.org/10.3390/cancers11050718
Chicago/Turabian StylePandey, Gaurav, Nicholas Borcherding, Ryan Kolb, Paige Kluz, Wei Li, Sonia Sugg, Jun Zhang, Dazhi A. Lai, and Weizhou Zhang. 2019. "ROR1 Potentiates FGFR Signaling in Basal-Like Breast Cancer" Cancers 11, no. 5: 718. https://doi.org/10.3390/cancers11050718
APA StylePandey, G., Borcherding, N., Kolb, R., Kluz, P., Li, W., Sugg, S., Zhang, J., Lai, D. A., & Zhang, W. (2019). ROR1 Potentiates FGFR Signaling in Basal-Like Breast Cancer. Cancers, 11(5), 718. https://doi.org/10.3390/cancers11050718