TMEM16F/Anoctamin 6 in Ferroptotic Cell Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
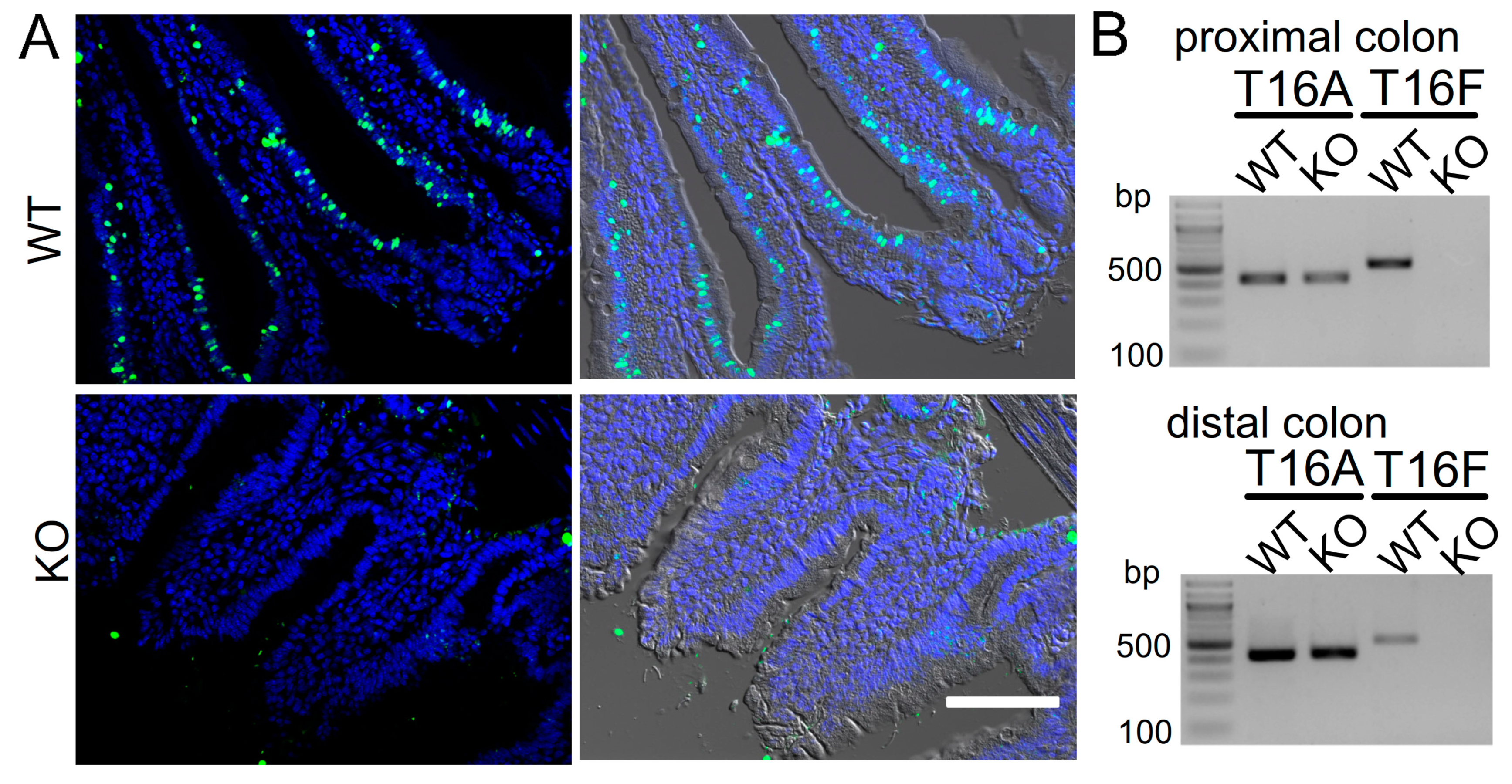
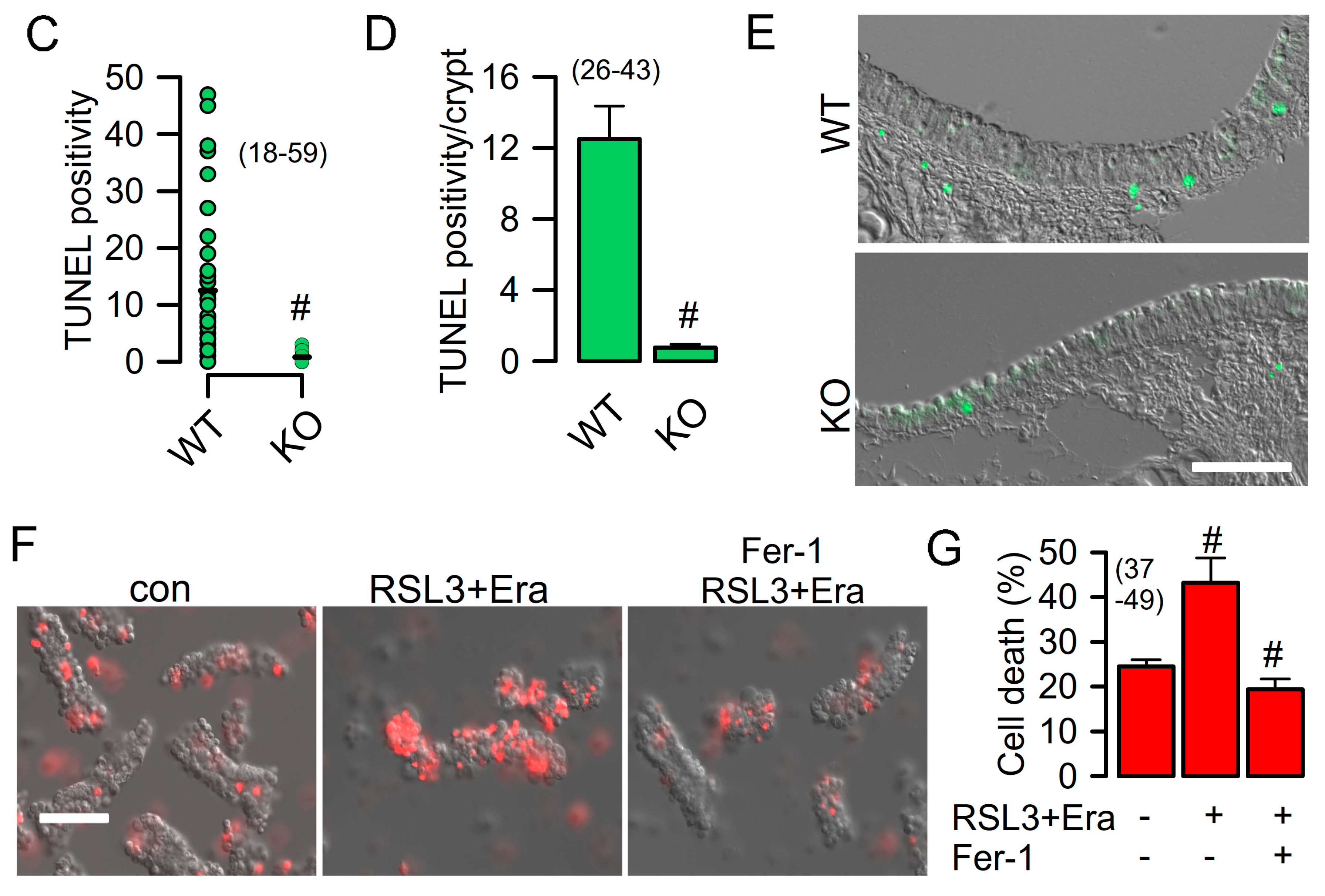
2.1. Attenuated Cell Death in Epithelial Cells from Mice Lacking Expression of TMEM16F
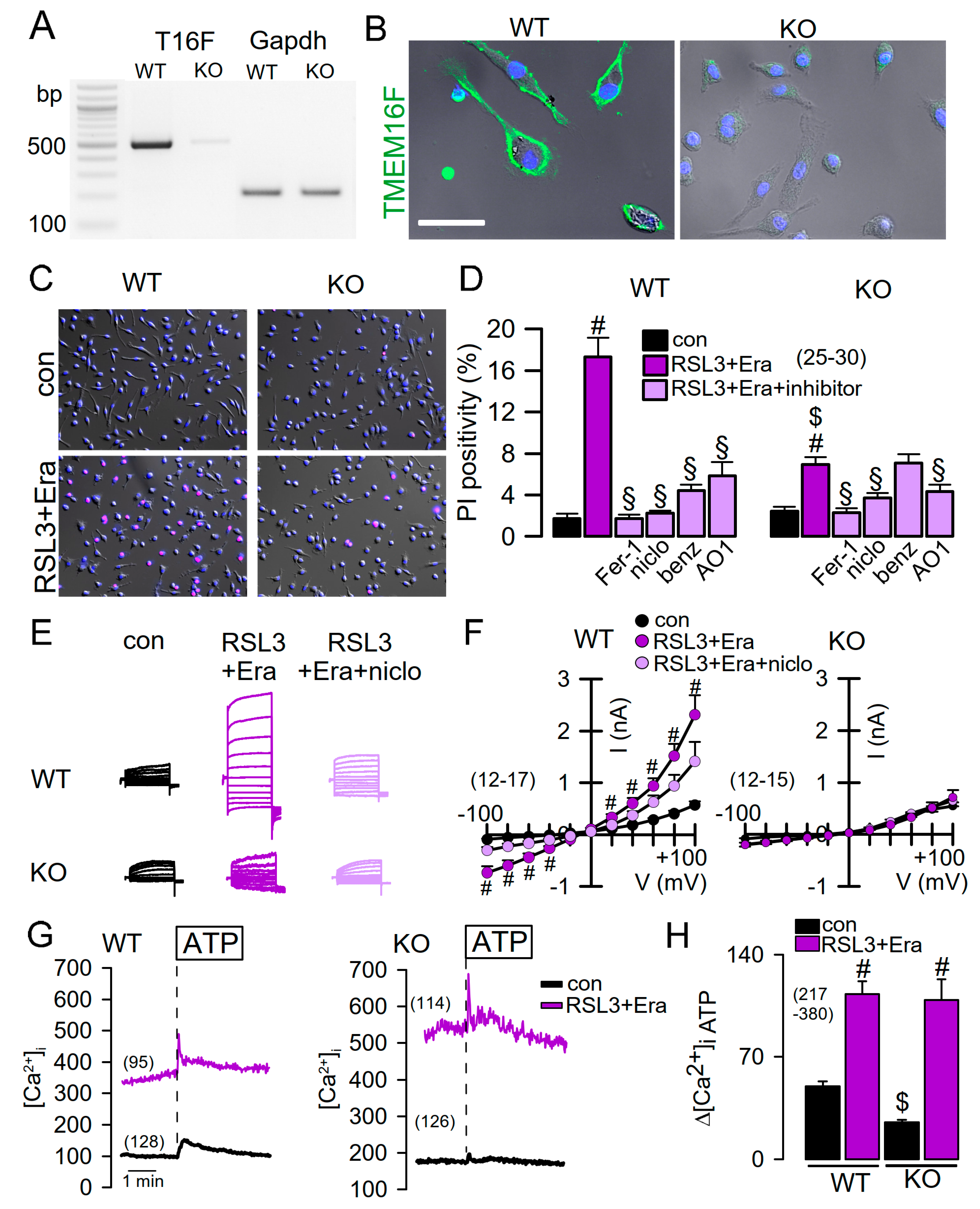
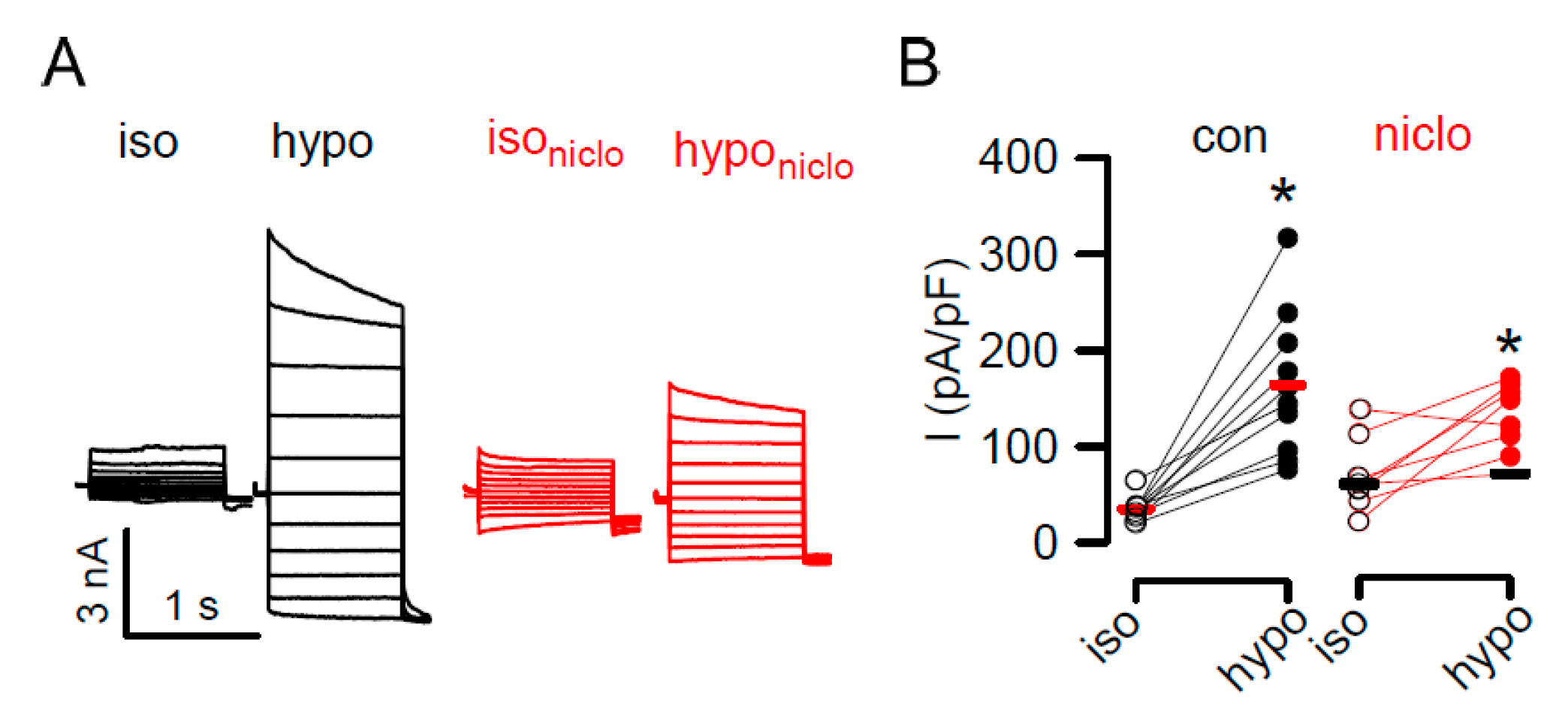
2.2. Reduced Ferroptosis in Macrophages from Mice Lacking Expression of TMEM16F
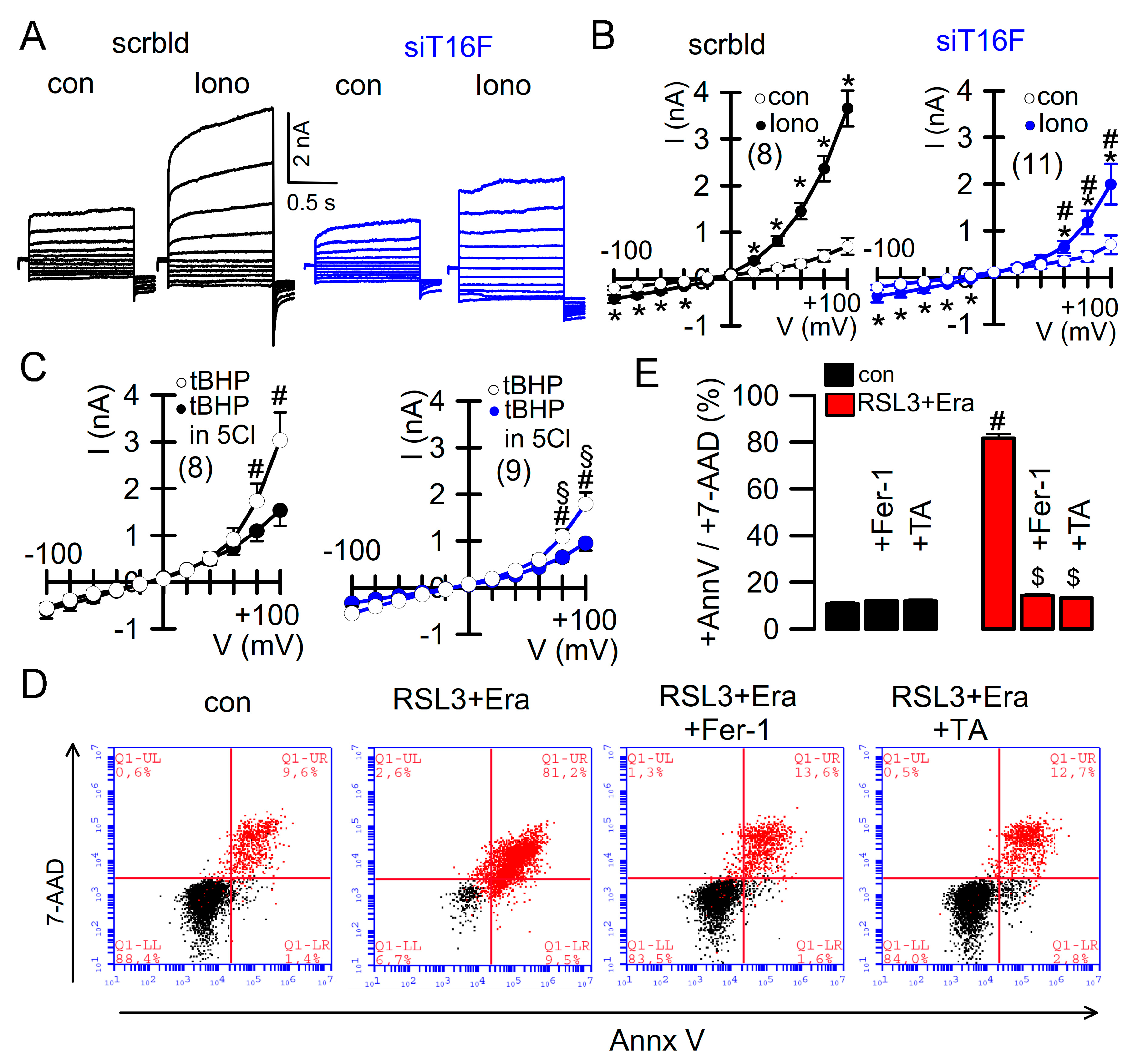
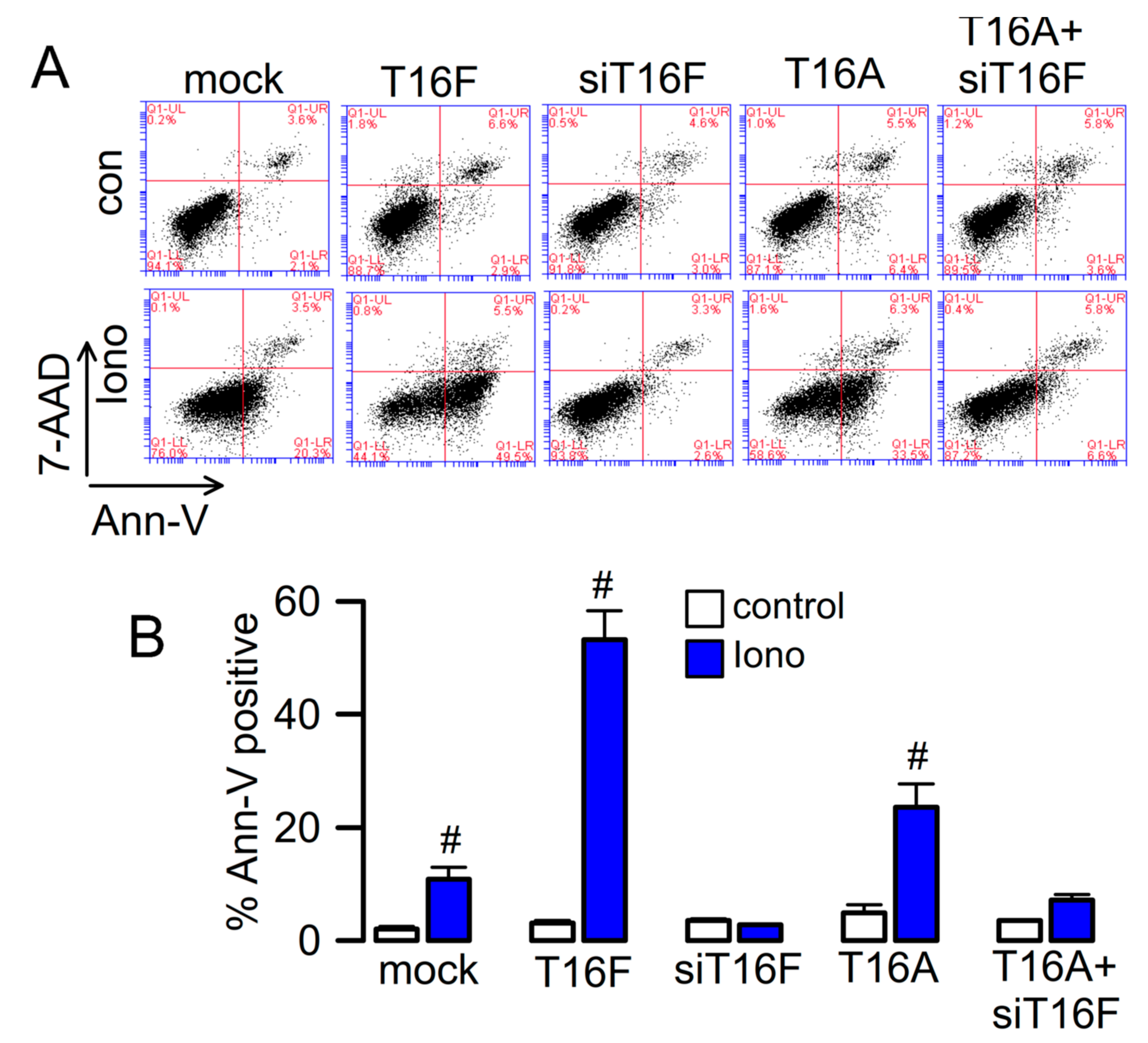
2.3. Effect of Overexpressed TMEM16F and Cooperativity with TMEM16A
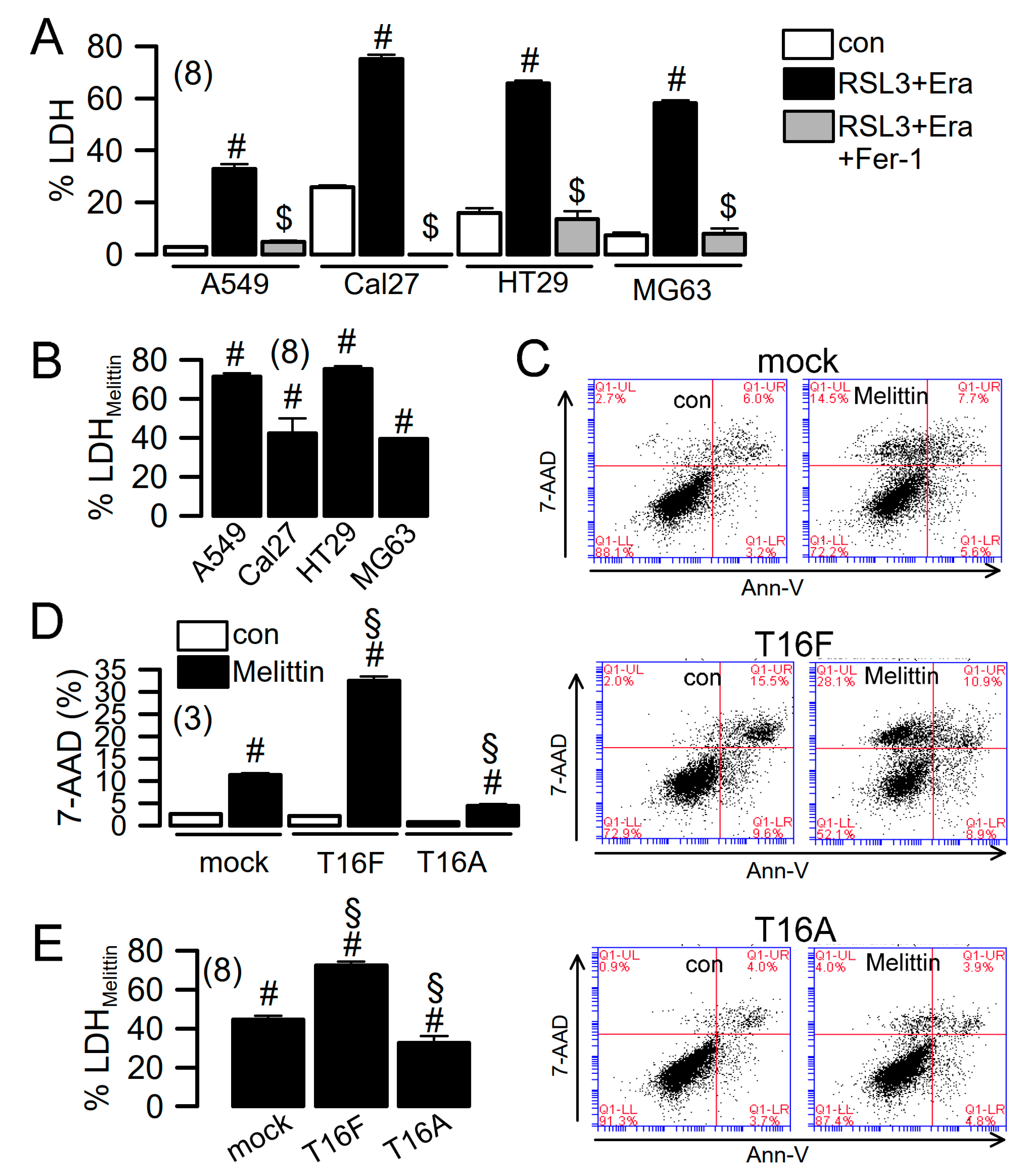
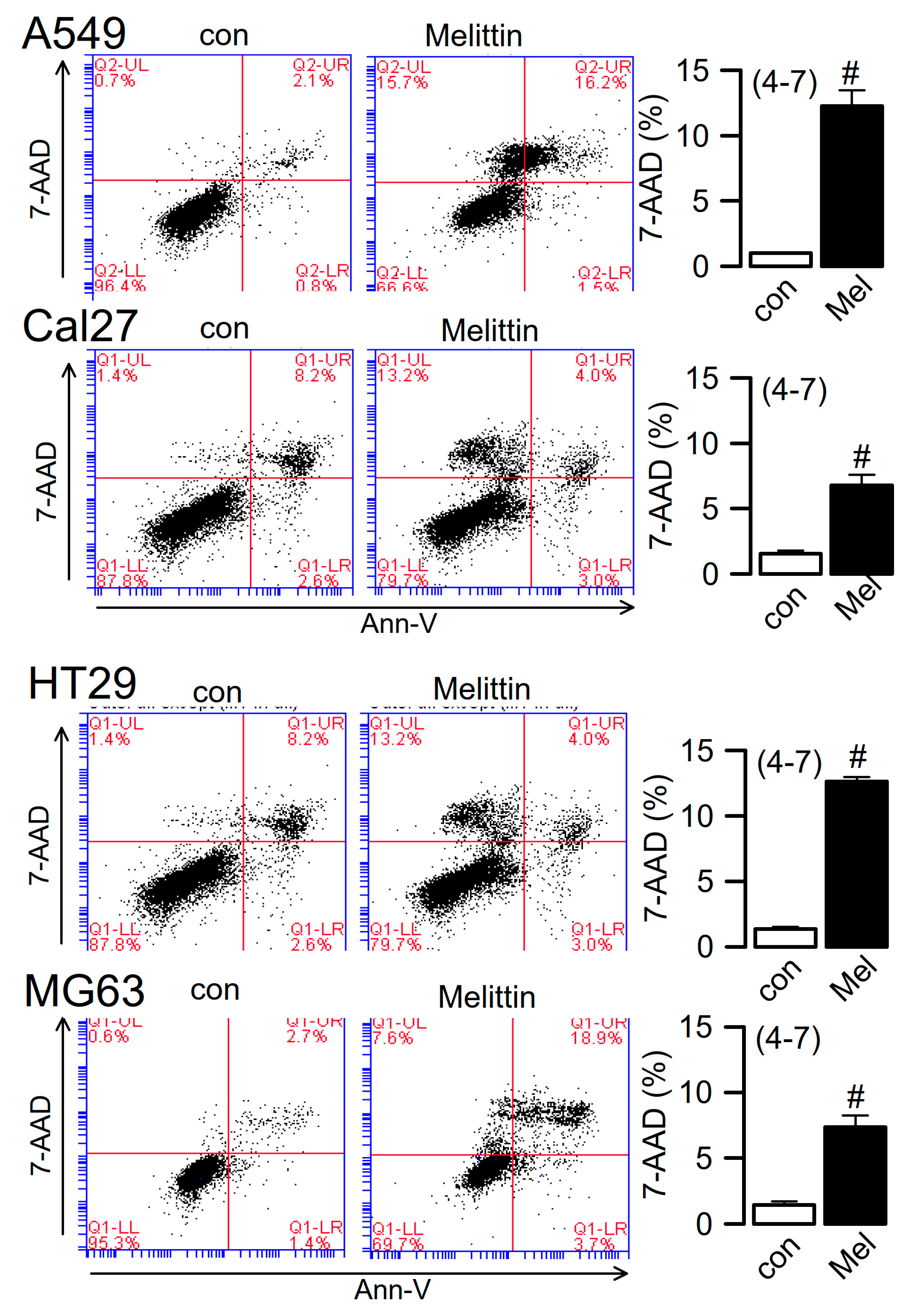
2.4. Ferroptosis Induced in Cancer Cells
3. Discussion
4. Methods
4.1. Immunocytochemistry
4.2. Isolation of Total RNA and RT-PCR
4.3. Calcium Measurements
4.4. Patch Clamping
4.5. TUNEL Assay
4.6. Flow Cytometry
4.7. LDH Assay
4.8. Knockout Animals
4.9. Material and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, A Membrane Protein Associated with Calcium-Dependent Chloride Channel Activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef]
- Wang, H.; Zou, L.; Ma, K.; Yu, J.; Wu, H.; Wei, M.; Xiao, Q. Cell-specific mechanisms of TMEM16A Ca2+-activated chloride channel in cancer. Mol. Cancer 2017, 16, 152. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Ousingsawat, J.; Benedetto, R.; Cabrita, I.; Schreiber, R. Growth and death by anoctamins. Cancers 2019, 19, E382. [Google Scholar] [CrossRef]
- Ruiz, C.; Martins, J.R.; Rudin, F.; Schneider, S.; Dietsche, T.; Fischer, C.A.; Tornillo, L.; Terracciano, L.M.; Schreiber, R.; Bubendorf, L.; et al. Enhanced Expression of ANO1 in Head and Neck Squamous Cell Carcinoma Causes Cell Migration and Correlates with Poor Prognosis. PLoS ONE 2012, 7, E43265. [Google Scholar] [CrossRef]
- Song, Y.; Gao, J.; Guan, L.; Chen, X.; Gao, J.; Wang, K. Inhibition of ANO1/TMEM16A induces apoptosis in human prostate carcinoma cells by activating TNF-alpha signaling. Cell Death Dis. 2018, 9, 703. [Google Scholar] [CrossRef]
- Seo, Y.; Park, J.; Kim, M.; Lee, H.K.; Kim, J.H.; Jeong, J.H.; Namkung, W. Inhibition of ANO1/TMEM16A Chloride Channel by Idebenone and Its Cytotoxicity to Cancer Cell Lines. PLoS ONE 2015, 10, E0133656. [Google Scholar] [CrossRef]
- Godse, N.R.; Khan, N.I.; Yochum, Z.A.; Gomez-Casal, R.; Kemp, C.; Shiwarski, D.J.; Seethala, R.; Kulich, S.; Seshadri, M.; Burns, T.F.; et al. TMEM16A/ANO1 inhibits apoptosis via down-regulation of Bim expression. Clin. Cancer Res. 2017, 23, 7324–7332. [Google Scholar] [CrossRef]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010, 468, 834–838. [Google Scholar] [CrossRef]
- Watanabe, R.; Sakuragi, T.; Noji, H.; Nagata, S. Single-molecule analysis of phospholipid scrambling by TMEM16F. Proc. Natl. Acad. Sci. USA 2018, 115, 3066–3071. [Google Scholar] [CrossRef]
- Brunner, J.D.; Lim, N.K.; Schenck, S.; Duerst, A.; Dutzler, R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 2014, 516, 207–212. [Google Scholar] [CrossRef]
- Yang, H.; Kim, A.; David, T.; Palmer, D.; Jin, T.; Tien, J.; Huang, F.; Cheng, T.; Coughlin, S.R.; Jan, Y.N.; et al. TMEM16F Forms a Ca2+-Activated Cation Channel Required for Lipid Scrambling in Platelets during Blood Coagulation. Cell 2012, 151, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Schreiber, R.; Kunzelmann, K. Anoctamins are a family of Ca2+ activated Cl− channels. J. Cell Sci. 2012, 125, 4991–4998. [Google Scholar] [CrossRef]
- Martins, J.R.; Faria, D.; Kongsuphol, P.; Reisch, B.; Schreiber, R.; Kunzelmann, K. Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc. Natl. Acad. Sci. USA 2011, 108, 18168–18172. [Google Scholar] [CrossRef]
- Shimizu, T.; Iehara, T.; Sato, K.; Fujii, T.; Sakai, H.; Okada, Y. TMEM16F is a component of a Ca2+-activated Cl− channel but not a volume-sensitive outwardly rectifying Cl− channel. Am. J. Physiol. Cell Physiol. 2013, 304, C748–C759. [Google Scholar] [CrossRef]
- Grubb, S.; Poulsen, K.A.; Juul, C.A.; Kyed, T.; Klausen, T.K.; Larsen, E.H.; Hoffmann, E.K. TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+ activation. J. Gen. Physiol. 2013, 141, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Whitlock, J.M.; Lee, K.; Ortlund, E.A.; Yuan, C.Y.; Hartzell, H.C. Identification of a lipid scrambling domain in ANO6/TMEM16F. eLife 2015, 4, e06901. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Wanitchakool, P.; Kmit, A.; Romao, A.M.; Jantarajit, W.; Schreiber, S.; Kunzelmann, K. Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7-receptors in macrophages. Nat. Commun. 2015, 6, 6245. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. (Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Contribution of TMEM16F to pyroptotic cell death. Cell Death Dis. 2018, 9, 300. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Ousingsawat, J.; Wanitchakool, P.; Sirianant, L.; Benedetto, R.; Reiss, K.; Kunzelmann, K. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca2+ and plasma membrane lipid. J. Physiol. 2018, 596, 217–229. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 2018, 24, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Muratori, C.; Pakhomov, A.G.; Gianulis, E.; Meads, J.; Casciola, M.; Mollica, P.A.; Pakhomova, O.N. Activation of the phospholipid scramblase TMEM16F by nanosecond pulsed electric field (nsPEF) facilitates its diverse cytophysiological effects. J. Biol. Chem. 2017, 292, 19381–19391. [Google Scholar] [CrossRef] [PubMed]
- Ousingsawat, J.; Cabrita, I.; Wanitchakool, P.; Sirianant, L.; Krautwald, S.; Linkermann, A.; Schreiber, R.; Kunzelmann, K. Ca2+ signals, cell membrane disintegration, and activation of TMEM16F during necroptosis. Cell. Mol. Life Sci. 2016, 74, 173–181. [Google Scholar] [CrossRef]
- Forschbach, V.; Goppelt-Struebe, M.; Kunzelmann, K.; Schreiber, R.; Piedagnel, R.; Kraus, A.; Eckardt, K.U.; Buchholz, B. Anoctamin 6 is localized in the primary cilium of renal tubular cells and is involved in apoptosis-dependent cyst lumen formation. Cell Death Dis. 2015, 6, E1899. [Google Scholar] [CrossRef] [PubMed]
- Simoes, F.; Ousingsawat, J.; Wanitchakool, P.; Fonseca, A.; Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. CFTR supports cell death through ROS-dependent activation of TMEM16F (anoctamin 6). Pflugers Arch. 2018, 470, 305–314. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Crosnier, C.; Stamataki, D.; Lewis, J. Organizing cell renewal in the intestine: Stem cells, signals and combinatorial control. Nat. Rev. Genet. 2006, 7, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. Niclosamide repurposed for the treatment of inflammatory airway disease. JCI Insight 2019. under review. [Google Scholar]
- Strater, J.; Wedding, U.; Barth, T.F.; Koretz, K.; Elsing, C.; Moller, P. Rapid onset of apoptosis in vitro follows disruption of beta 1-integrin/matrix interactions in human colonic crypt cells. Gastroenterology 1996, 110, 1776–1784. [Google Scholar] [CrossRef]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Sirianant, L.; Ousingsawat, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Cellular Volume regulation by Anoctamin 6: Ca2+, phospholipase A2,osmosensing. Pflügers Arch. 2015, 468, 335–349. [Google Scholar]
- Kunzelmann, K.; Nilius, B.; Owsianik, G.; Schreiber, R.; Ousingsawat, J.; Sirianant, L.; Wanitchakool, P.; Bevers, E.M.; Heemskerk, J.W. Molecular functions of anoctamin 6 (TMEM16F): A. chloride channel, cation channel or phospholipid scramblase? Pflügers Arch. 2014, 466, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Gyobu, S.; Ishihara, K.; Suzuki, J.; Segawa, K.; Nagata, S. Characterization of the scrambling domain of the TMEM16 family. Proc. Natl. Acad. Sci. USA 2017, 114, 6274–6279. [Google Scholar] [CrossRef]
- Cabrita, I.; Benedetto, R.; Fonseca, A.; Wanitchakool, P.; Sirianant, L.; Skryabin, B.V.; Schenk, L.K.; Pavenstadt, H.; Schreiber, R.; Kunzelmann, K. Differential effects of anoctamins on intracellular calcium signals. FASEB J. 2017, 31, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Uliyakina, I.; Kongsuphol, P.; Warth, R.; Mirza, M.; Martins, J.R.; Kunzelmann, K. Expression and Function of Epithelial Anoctamins. J. Biol. Chem. 2010, 285, 7838–7845. [Google Scholar] [CrossRef]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 130, 126428. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed]
- Alvadia, C.; Lim, N.K.; Clerico Mosina, V.; Oostergetel, G.T.; Dutzler, R.; Paulino, C. Cryo-EM structures and functional characterization of the murine lipid scramblase TMEM16F. eLife 2019, 8, e44365. [Google Scholar] [CrossRef]
- Paulino, C.; Kalienkova, V.; Lam, A.K.M.; Neldner, Y.; Dutzler, R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 2017, 552, 421–425. [Google Scholar] [CrossRef]
- Dang, S.; Feng, S.; Tien, J.; Peters, C.J.; Bulkley, D.; Lolicato, M.; Zhao, J.; Zuberbuhler, K.; Ye, W.; Qi, L.; et al. Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature 2017, 552, 426. [Google Scholar] [CrossRef]
- Kalienkova, V.; Clerico Mosina, V.; Bryner, L.; Oostergetel, G.T.; Dutzler, R.; Paulino, C. Stepwise activation mechanism of the scramblase nhTMEM16 revealed by cryo-EM. eLife 2019, 8, e44364. [Google Scholar] [CrossRef]
- Tian, Y.; Schreiber, R.; Wanitchakool, P.; Kongsuphol, P.; Sousa, M.; Uliyakina, I.; Palma, M.; Faria, D.; Traynor-Kaplen, A.E.; Fragata, J.I.; et al. Control of TMEM16A by INO-4995 and other inositolphosphates. Br. J. Pharmacol. 2012, 168, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Oh, U. Anoctamin 1 mediates thermal pain as a heat sensor. Curr. Neuropharmacol. 2013, 11, 641–651. [Google Scholar] [CrossRef]
- Mattheij, N.J.; Braun, A.; van Kruchten, R.; Castoldi, E.; Pircher, J.; Baaten, C.C.; Wulling, M.; Kuijpers, M.J.; Kohler, R.; Poole, A.W.; et al. Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling, and protein cleavage. FASEB J. 2015, 30, 727–737. [Google Scholar] [CrossRef]
- Liu, G.; Liu, G.; Chen, H.; Borst, O.; Gawaz, M.; Vortkamp, A.; Schreiber, R.; Kunzelmann, K.; Lang, F. Involvement of Ca2+ Activated Cl− Channel Ano6 in Platelet Activation and Apoptosis. Cell Physiol. Biochem. 2015, 37, 1934–1944. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Rad. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Bricogne, C.; Fine, M.; Pereira, P.M.; Sung, J.; Tijani, M.; Wang, Y.; Henriques, R.; Collins, M.K.; Hilgemann, D. TMEM16F activation by Ca2+ triggers plasma membrane expansion and directs PD-1 trafficking. Sci. Rep. 2019, 9, 619. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, R.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019, 33, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, R.; Ousingsawat, J.; Cabrita, I.; Pinto, M.; Lerias, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Plasma membrane localized TMEM16 Proteins are Indispensable for expression of CFTR. J. Mol. Med. 2019, 97, 711–722. [Google Scholar] [CrossRef]
- Benedetto, R.; Sirianant, L.; Pankonien, I.; Wanitchakool, P.; Ousingsawat, J.; Cabrita, I.; Schreiber, R.; Amaral, M.; Kunzelmann, K. Relationship between TMEM16A/anoctamin 1 and LRRC8A. Pflugers Arch. 2016, 468, 1751–1763. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, Z.; Zhang, D.; Li, H.; Zhang, L.; Niu, J.; Zuo, W.; Fu, R.; Fan, L.; Ye, J.H.; et al. High expression of leucinerich repeatcontaining 8A is indicative of a worse outcome of colon cancer patients by enhancing cancer cell growth and metastasis. Oncology Rep. 2018, 40, 1275–1286. [Google Scholar] [CrossRef]
- Almaca, J.; Tian, Y.; AlDehni, F.; Ousingsawat, J.; Kongsuphol, P.; Rock, J.R.; Harfe, B.D.; Schreiber, R.; Kunzelmann, K. TMEM16 proteins produce volume regulated chloride currents that are reduced in mice lacking TMEM16A. J. Biol. Chem. 2009, 284, 28571–28578. [Google Scholar] [CrossRef]
- Miner, K.; Labitzke, K.; Liu, B.; Elliot, R.; Wang, P.; Henckels, K.; Gaida, K.; Elliot, R.; Chen, J.J.; Liu, L.; et al. The Anthelminthic Niclosamide And Related Compounds Represent Potent Tmem16a Antagonists That Fully Relax Mouse and Human Airway Rings. Frontiers Pharmacol. 2019, 14. [Google Scholar] [CrossRef]
- Zhao, P.; Torcaso, A.; Mariano, A.; Xu, L.; Mohsin, S.; Zhao, L.; Han, R. Anoctamin 6 Regulates C2C12 Myoblast Proliferation. PLoS ONE 2014, 9, E92749. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers 2019, 11, 625. https://doi.org/10.3390/cancers11050625
Ousingsawat J, Schreiber R, Kunzelmann K. TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers. 2019; 11(5):625. https://doi.org/10.3390/cancers11050625
Chicago/Turabian StyleOusingsawat, Jiraporn, Rainer Schreiber, and Karl Kunzelmann. 2019. "TMEM16F/Anoctamin 6 in Ferroptotic Cell Death" Cancers 11, no. 5: 625. https://doi.org/10.3390/cancers11050625
APA StyleOusingsawat, J., Schreiber, R., & Kunzelmann, K. (2019). TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers, 11(5), 625. https://doi.org/10.3390/cancers11050625