Influence of the HDAC Inhibitor Valproic Acid on the Growth and Proliferation of Temsirolimus-Resistant Prostate Cancer Cells In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs
2.3. Tumor Cell Growth and Proliferation
2.4. Clonogenic Growth Assay
2.5. Cell Cycle Analysis
2.6. Western Blot Analysis
2.7. siRNA Knockdown
2.8. Statistics
3. Results
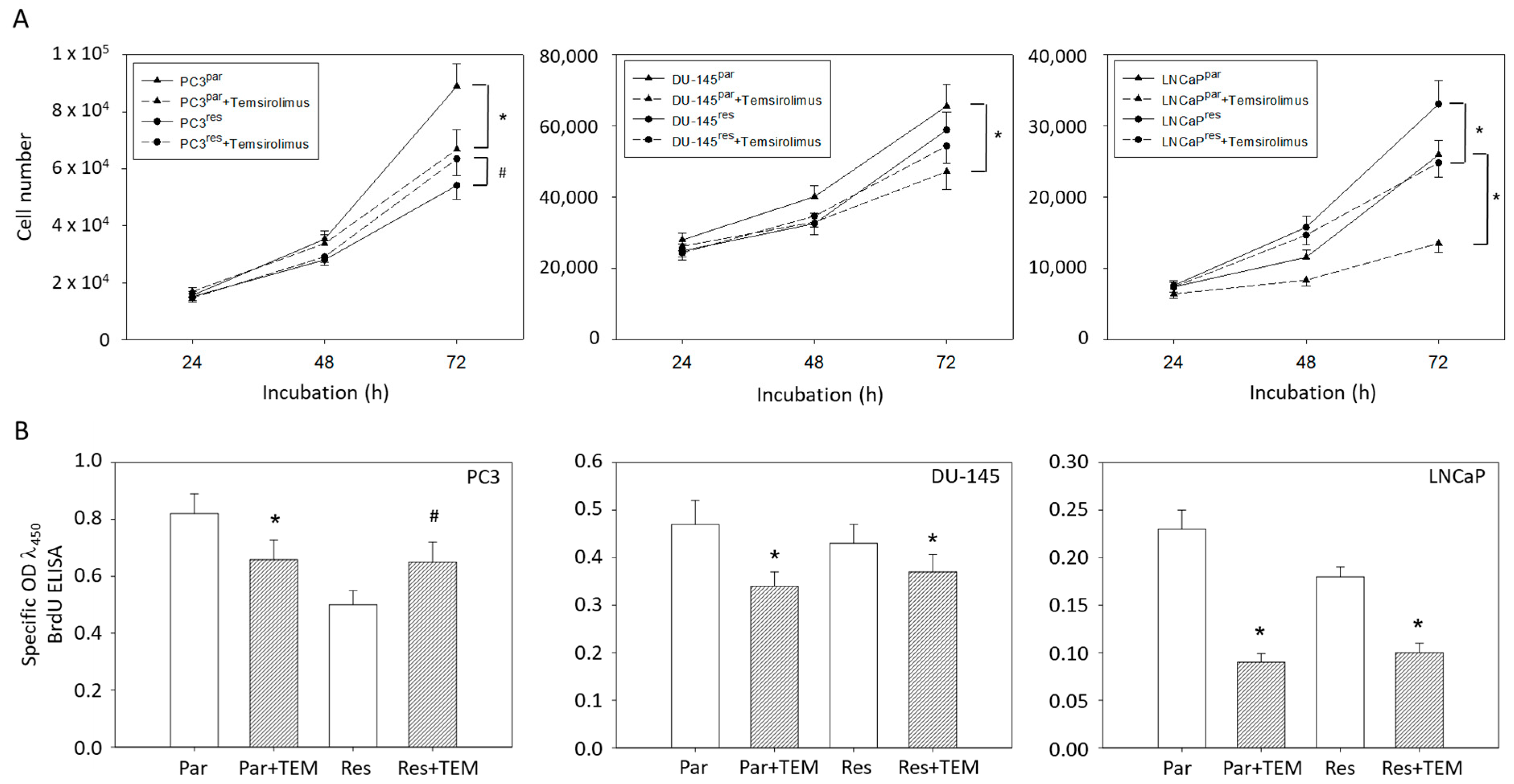
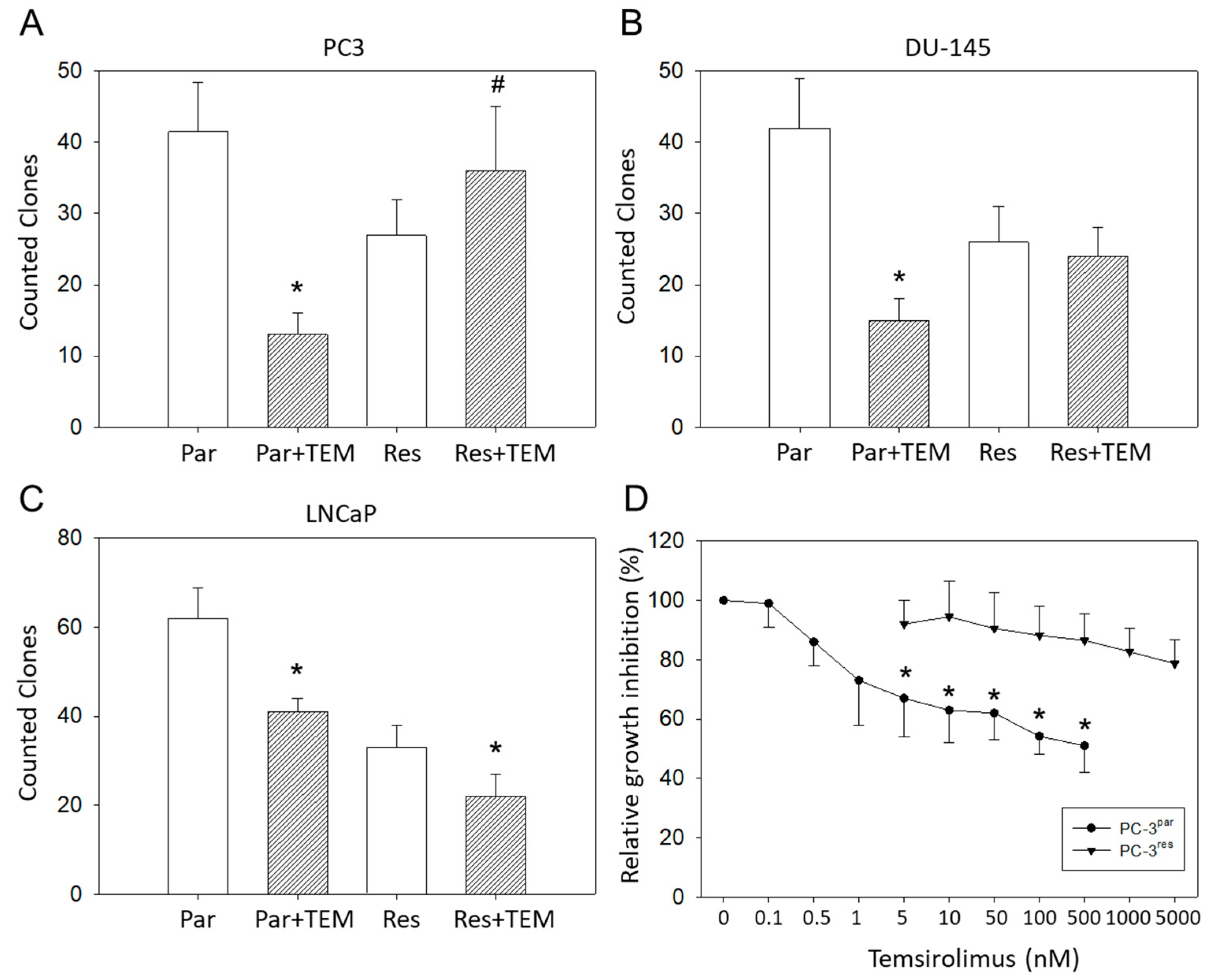
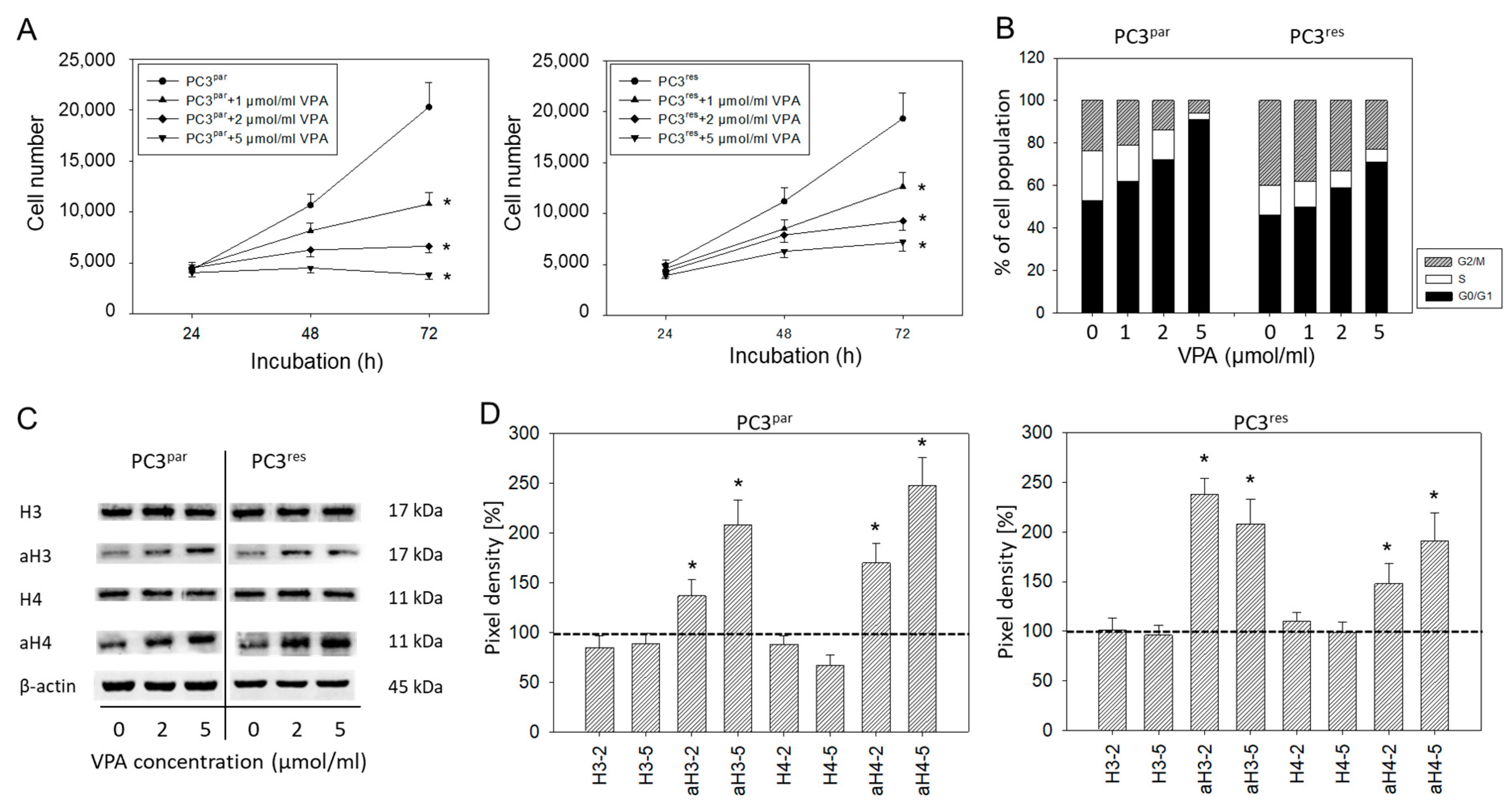
3.1. Tumor Growth and Proliferation
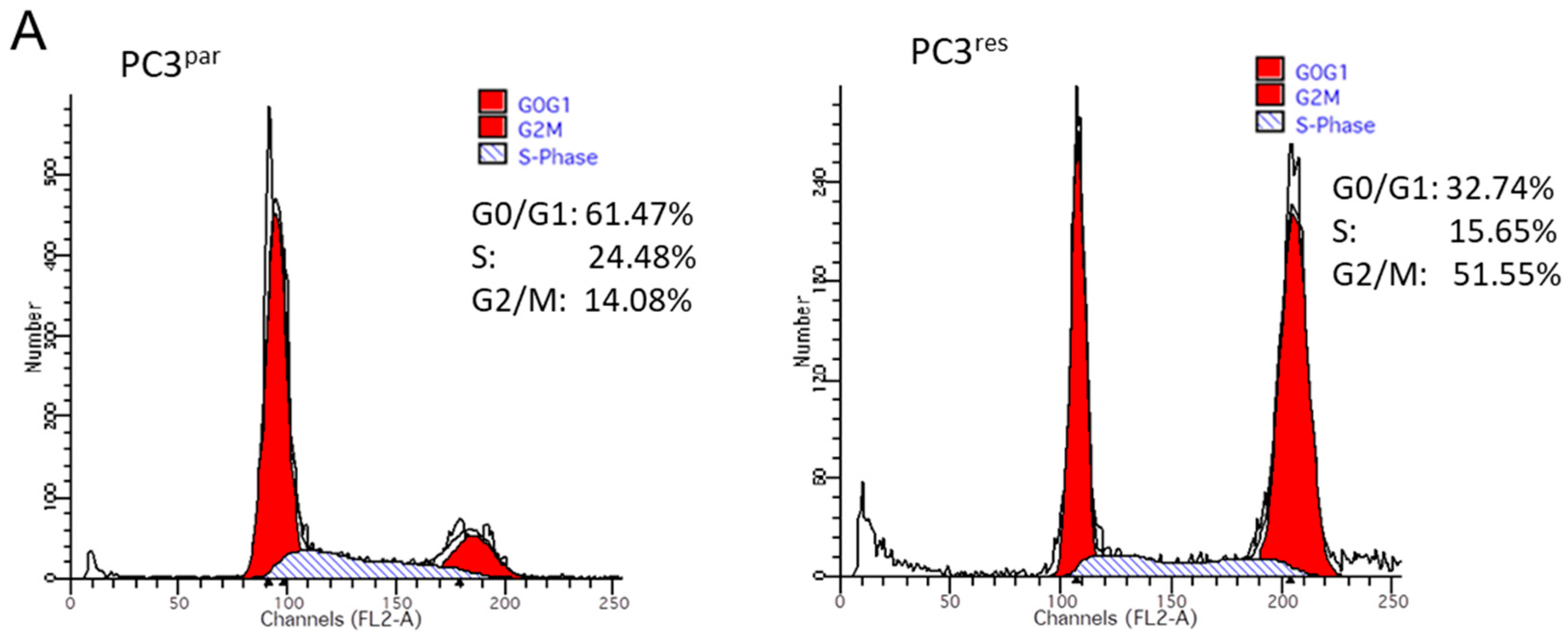
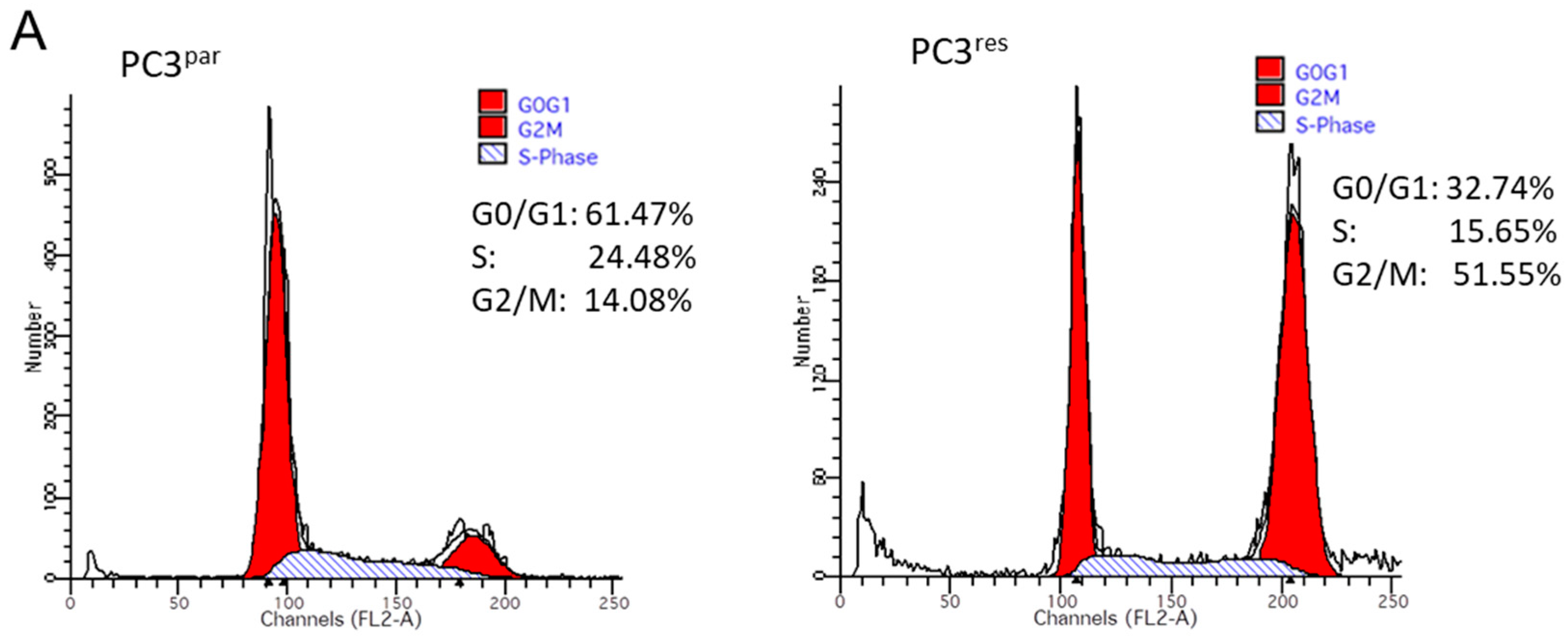
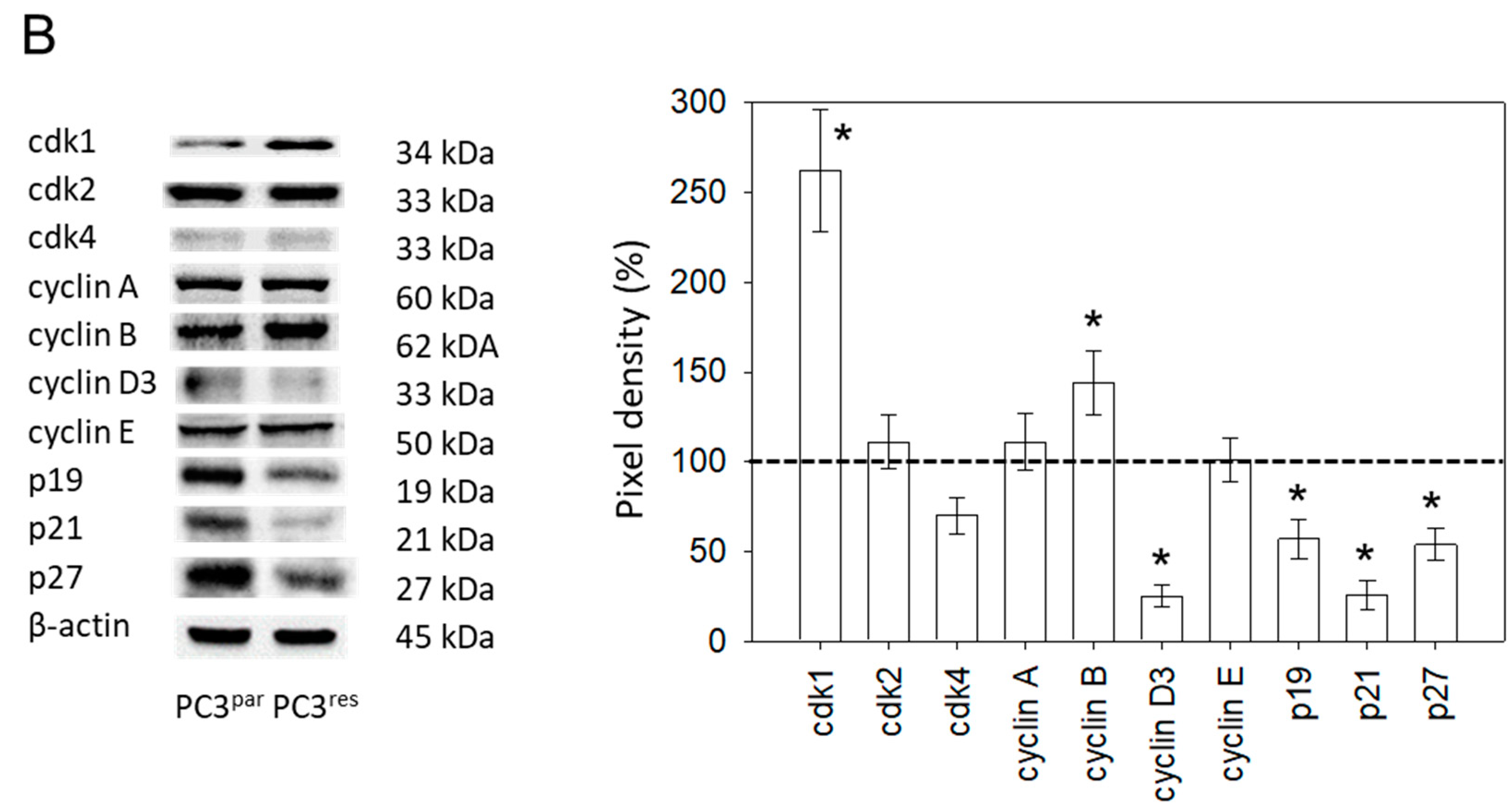
3.2. Cell Cycle Analysis and Cell Cycle-Regulating Protein Expression
3.3. VPA Suppresses Growth of PC3par and PC3res Cells
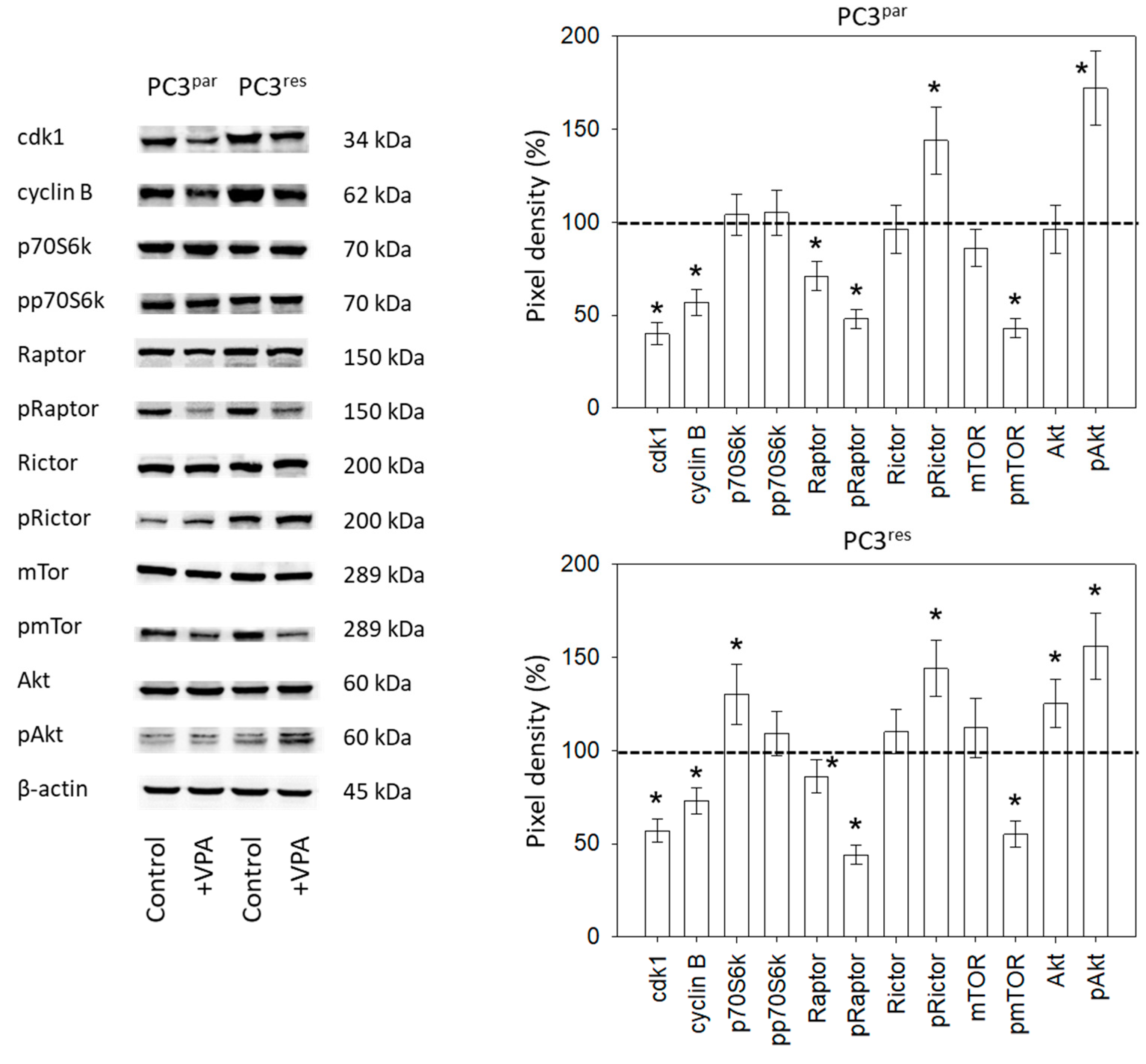
3.4. Influence of VPA on Cell Signaling Processes
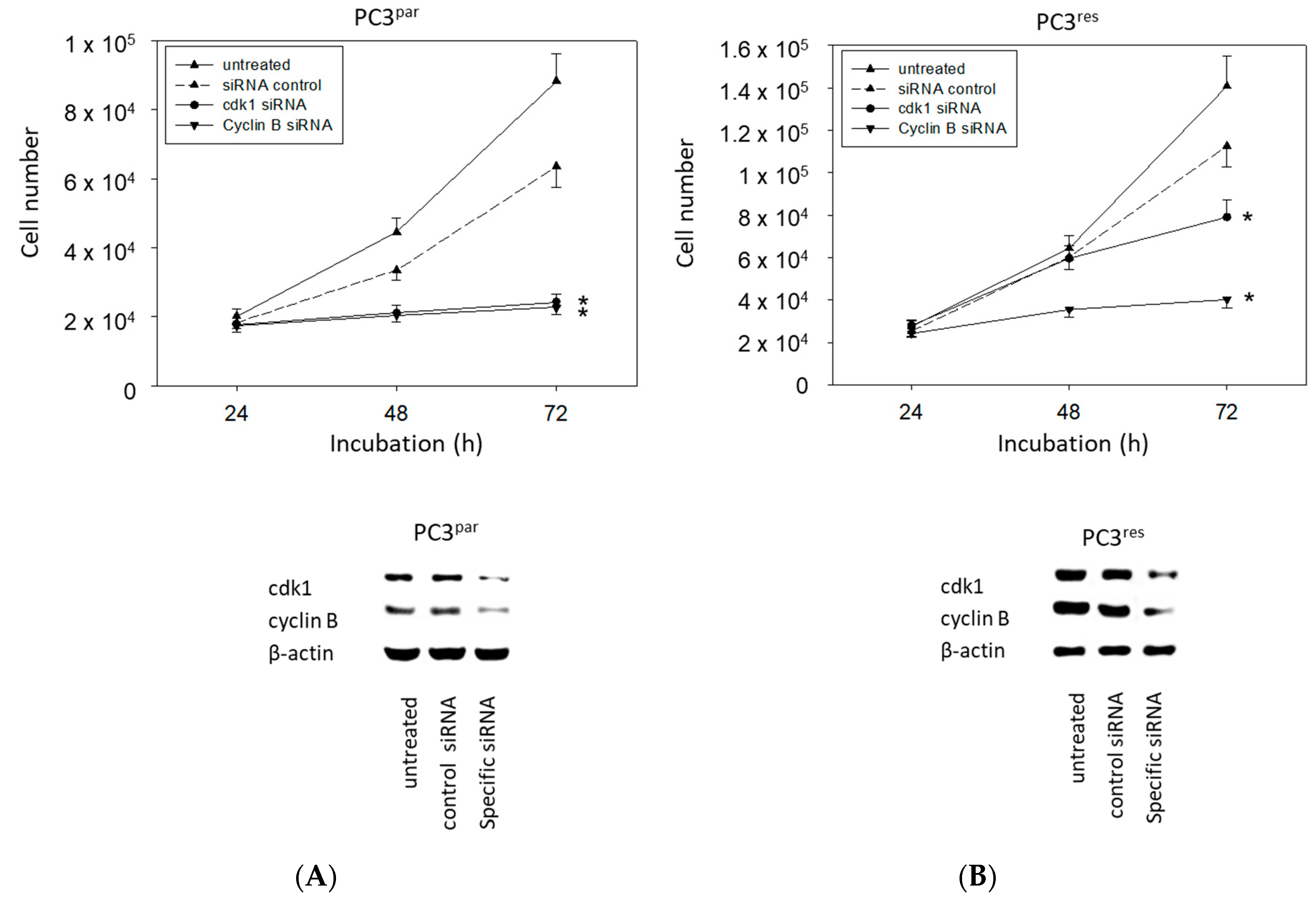
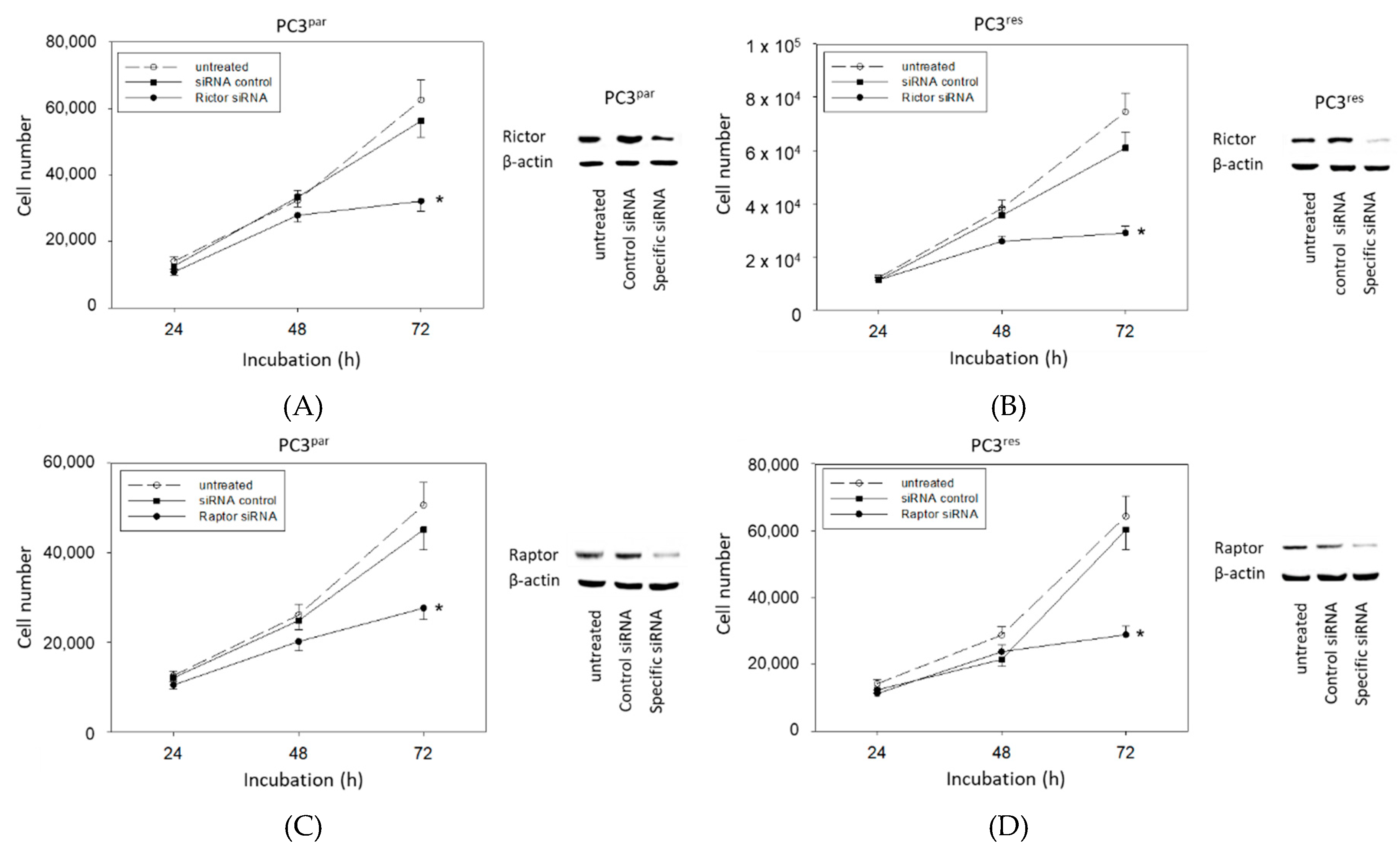
3.5. Protein Knockdown Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dybos, S.A.; Flatberg, A.; Halgunset, J.; Viset, T.; Rolfseng, T.; Kvam, S.; Skogseth, H. Increased levels of serum miR-148a-3p are associated with prostate cancer. APMIS 2018, 126, 722–731. [Google Scholar] [CrossRef]
- Ryan, C.J.; Tindall, D.J. Androgen receptor rediscovered: The new biology and targeting the androgen receptor therapeutically. J. Clin. Oncol. 2011, 29, 3651–3658. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Shen, W.; He, P.; Zhou, Z. Effectiveness and tolerability of targeted drugs for the treatment of metastatic castration-resistant prostate cancer: A network meta-analysis of randomized controlled trials. J. Cancer Res. Clin. Oncol. 2018, 144, 1751–1768. [Google Scholar] [CrossRef] [PubMed]
- Summers, N.; Vanderpuye-Orgle, J.; Reinhart, M.; Gallagher, M.; Sartor, O. Efficacy and safety of post-docetaxel therapies in metastatic castration-resistant prostate cancer: A systematic review of the literature. Curr. Med. Res. Opin. 2017, 33, 1995–2008. [Google Scholar] [CrossRef]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Kato, M.; Banuelos, C.A.; Imamura, Y.; Leung, J.K.; Caley, D.P.; Wang, J.; Mawji, N.R.; Sadar, M.D. Cotargeting Androgen Receptor Splice Variants and mTOR Signaling Pathway for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 2744–2754. [Google Scholar] [CrossRef]
- de Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L.; et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Invest. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef]
- Earwaker, P.; Anderson, C.; Willenbrock, F.; Harris, A.L.; Protheroe, A.S.; Macaulay, V.M. RAPTOR up-regulation contributes to resistance of renal cancer cells to PI3K-mTOR inhibition. PLoS ONE 2018, 13, e0191890. [Google Scholar] [CrossRef]
- Juengel, E.; Dauselt, A.; Makarević, J.; Wiesner, C.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal cell carcinoma cells. Cancer Lett. 2012, 324, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Makarević, J.; Tawanaie, N.; Juengel, E.; Reiter, M.; Mani, J.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Cross-communication between histone H3 and H4 acetylation and Akt-mTOR signalling in prostate cancer cells. J. Cell Mol. Med. 2014, 18, 1460–1466. [Google Scholar] [CrossRef]
- Park, H.; Garrido-Laguna, I.; Naing, A.; Fu, S.; Falchook, G.S.; Piha-Paul, S.A.; Wheler, J.J.; Hong, D.S.; Tsimberidou, A.M.; Subbiah, V.; et al. Phase I dose-escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget 2016, 7, 67521–67531. [Google Scholar] [CrossRef]
- Juengel, E.; Najafi, R.; Rutz, J.; Maxeiner, S.; Makarevic, J.; Roos, F.; Tsaur, I.; Haferkamp, A.; Blaheta, R.A. HDAC inhibition as a treatment concept to combat temsirolimus-resistant bladder cancer cells. Oncotarget 2017, 8, 110016–110028. [Google Scholar] [CrossRef]
- Juengel, E.; Nowaz, S.; Makarevi, J.; Natsheh, I.; Werner, I.; Nelson, K.; Reiter, M.; Tsaur, I.; Mani, J.; Harder, S.; et al. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol. Cancer 2014, 13, 152. [Google Scholar] [CrossRef]
- Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Mani, J.; Vallo, S.; Tsaur, I.; Roos, F.; Chun, F.K.; Blaheta, R.A. HDAC Inhibition Counteracts Metastatic Re-Activation of Prostate Cancer Cells Induced by Chronic mTOR Suppression. Cells 2018, 7, 129. [Google Scholar] [CrossRef]
- Tsaur, I.; Makarević, J.; Hudak, L.; Juengel, E.; Kurosch, M.; Wiesner, C.; Bartsch, G.; Harder, S.; Haferkamp, A.; Blaheta, R.A. The cdk1-cyclin B complex is involved in everolimus triggered resistance in the PC3 prostate cancer cell line. Cancer Lett. 2011, 313, 84–90. [Google Scholar] [CrossRef]
- Yamada, Y.; Nishikawa, R.; Kato, M.; Okato, A.; Arai, T.; Kojima, S.; Yamazaki, K.; Naya, Y.; Ichikawa, T.; Seki, N. Regulation of HMGB3 by antitumor miR-205-5p inhibits cancer cell aggressiveness and is involved in prostate cancer pathogenesis. J. Hum. Genet. 2018, 63, 195–205. [Google Scholar] [CrossRef]
- Goto, Y.; Kurozumi, A.; Arai, T.; Nohata, N.; Kojima, S.; Okato, A.; Kato, M.; Yamazaki, K.; Ishida, Y.; Naya, Y.; et al. Impact of novel miR-145-3p regulatory networks on survival in patients with castration-resistant prostate cancer. Br. J. Cancer 2017, 117, 409–420. [Google Scholar] [CrossRef]
- Giesing, M.; Driesel, G.; Molitor, D.; Suchy, B. Molecular phenotyping of circulating tumour cells in patients with prostate cancer: Prediction of distant metastases. BJU Int. 2012, 110, e1202–e1211. [Google Scholar] [CrossRef]
- Chen, G.; Yang, N.; Wang, X.; Zheng, S.Y.; Chen, Y.; Tong, L.J.; Li, Y.X.; Meng, L.H.; Ding, J. Identification of p27/KIP1 expression level as a candidate biomarker of response to rapalogs therapy in human cancer. J. Mol. Med. 2010, 88, 941–952. [Google Scholar] [CrossRef]
- Singh, S.K.; Banerjee, S.; Acosta, E.P.; Lillard, J.W.; Singh, R. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and p27KIP1 pathway. Oncotarget 2017, 8, 17216–17228. [Google Scholar] [CrossRef]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef]
- Wang, C.; Sun, H.; Zou, R.; Zhou, T.; Wang, S.; Sun, S.; Tong, C.; Luo, H.; Li, Y.; Li, Z.; et al. MDC1 functionally identified as an androgen receptor co-activator participates in suppression of prostate cancer. Nucleic Acids Res. 2015, 43, 4893–4908. [Google Scholar] [CrossRef]
- Sociali, G.; Galeno, L.; Parenti, M.D.; Grozio, A.; Bauer, I.; Passalacqua, M.; Boero, S.; Donadini, A.; Millo, E.; Bellotti, M.; et al. Quinazolinedione SIRT6 inhibitors sensitize cancer cells to chemotherapeutics. Eur. J. Med. Chem. 2015, 102, 530–539. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, R.; Su, Z.Y.; Guo, Y.; Zheng, X.; Yang, C.S.; Kong, A.N. A naturally occurring mixture of tocotrienols inhibits the growth of human prostate tumor, associated with epigenetic modifications of cyclin-dependent kinase inhibitors p21 and p27. J. Nutr. Biochem. 2017, 40, 155–163. [Google Scholar] [CrossRef]
- Sajadpoor, Z.; Amini-Farsani, Z.; Teimori, H.; Shamsara, M.; Sangtarash, M.H.; Ghasemi-Dehkordi, P.; Yadollahi, F. Valproic Acid Promotes Apoptosis and Cisplatin Sensitivity Through Downregulation of H19 Noncoding RNA in Ovarian A2780 Cells. Appl. Biochem. Biotechnol. 2018, 185, 1132–1144. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhang, L.; Zhu, Y.; Xie, Q.; Zhu, B.; Chen, Z. Valproic acid, an inhibitor of class I histone deacetylases, reverses acquired Erlotinib-resistance of lung adenocarcinoma cells: A Connectivity Mapping analysis and an experimental study. Am. J. Cancer Res. 2015, 5, 2202–2211. [Google Scholar]
- Matsuda, Y.; Wakai, T.; Kubota, M.; Osawa, M.; Hirose, Y.; Sakata, J.; Kobayashi, T.; Fujimaki, S.; Takamura, M.; Yamagiwa, S.; et al. Valproic acid overcomes transforming growth factor-β-mediated sorafenib resistance in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 1299–1313. [Google Scholar]
- Wang, Y.; Kuramitsu, Y.; Kitagawa, T.; Tokuda, K.; Baron, B.; Akada, J.; Nakamura, K. The Histone Deacetylase Inhibitor Valproic Acid Sensitizes Gemcitabine-Induced Cytotoxicity in Gemcitabine-Resistant Pancreatic Cancer Cells Possibly Through Inhibition of the DNA Repair Protein Gamma-H2AX. Target. Oncol. 2015, 10, 575–581. [Google Scholar] [CrossRef]
- Blaheta, R.A.; Michaelis, M.; Natsheh, I.; Hasenberg, C.; Weich, E.; Relja, B.; Jonas, D.; Doerr, H.W.; Cinatl, J., Jr. Valproic acid inhibits adhesion of vincristine- and cisplatin-resistant neuroblastoma tumour cells to endothelium. Br. J. Cancer 2007, 96, 1699–1706. [Google Scholar] [CrossRef]
- Velásquez, C.; Cheng, E.; Shuda, M.; Lee-Oesterreich, P.J.; Pogge von Strandmann, L.; Gritsenko, M.A.; Jacobs, J.M.; Moore, P.S.; Chang, Y. Mitotic protein kinase CDK1 phosphorylation of mRNA translation regulator 4E-BP1 Ser83 may contribute to cell transformation. Proc. Natl. Acad. Sci. USA 2016, 113, 8466–8471. [Google Scholar] [CrossRef]
- Li, A.; Fan, S.; Xu, Y.; Meng, J.; Shen, X.; Mao, J.; Zhang, L.; Zhang, X.; Moeckel, G.; Wu, D.; et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J. Cell Mol. Med. 2017, 21, 1619–1635. [Google Scholar] [CrossRef]
- Li, N.; Xue, W.; Yuan, H.; Dong, B.; Ding, Y.; Liu, Y.; Jiang, M.; Kan, S.; Sun, T.; Ren, J.; et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Invest. 2017, 127, 1284–1302. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef]
- Oneyama, C.; Kito, Y.; Asai, R.; Ikeda, J.; Yoshida, T.; Okuzaki, D.; Kokuda, R.; Kakumoto, K.; Takayama, K.; Inoue, S.; et al. MiR-424/503-mediated Rictor upregulation promotes tumor progression. PLoS ONE 2013, 8, e80300. [Google Scholar] [CrossRef]
- Xia, Q.; Zheng, Y.; Jiang, W.; Huang, Z.; Wang, M.; Rodriguez, R.; Jin, X. Valproic acid induces autophagy by suppressing the Akt/mTOR pathway in human prostate cancer cells. Oncol. Lett. 2016, 12, 1826–1832. [Google Scholar] [CrossRef]
- Kotajima-Murakami, H.; Kobayashi, T.; Kashii, H.; Sato, A.; Hagino, Y.; Tanaka, M.; Nishito, Y.; Takamatsu, Y.; Uchino, S.; Ikeda, K. Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero. Mol. Brain 2019, 12, 3. [Google Scholar] [CrossRef]
- Lima, I.V.A.; Almeida-Santos, A.F.; Ferreira-Vieira, T.H.; Aguiar, D.C.; Ribeiro, F.M.; Campos, A.C.; de Oliveira, A.C.P. Antidepressant-like effect of valproic acid-Possible involvement of PI3K/Akt/mTOR pathway. Behav. Brain Res. 2017, 329, 166–171. [Google Scholar] [CrossRef]
- Juengel, E.; Makarević, J.; Tsaur, I.; Bartsch, G.; Nelson, K.; Haferkamp, A.; Blaheta, R.A. Resistance after chronic application of the HDAC-inhibitor valproic acid is associated with elevated Akt activation in renal cell carcinoma in vivo. PLoS ONE 2013, 8, e53100. [Google Scholar] [CrossRef]
- Salminen, J.K.; Tammela, T.L.; Auvinen, A.; Murtola, T.J. Antiepileptic drugs with histone deacetylase inhibition activity and prostate cancer risk: Apopulation-based case-control study. Cancer Causes Control. 2016, 27, 637–645. [Google Scholar] [CrossRef]
- Kaushik, D.; Vashistha, V.; Isharwal, S.; Sediqe, S.A.; Lin, M.F. Histone deacetylase inhibitors in castration-resistant prostate cancer: Molecular mechanism of action and recent clinical trials. Ther. Adv. Urol. 2015, 7, 388–395. [Google Scholar] [CrossRef]
- Eigl, B.J.; North, S.; Winquist, E.; Finch, D.; Wood, L.; Sridhar, S.S.; Powers, J.; Good, J.; Sharma, M.; Squire, J.A.; et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Invest. New Drugs 2015, 33, 969–976. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef]
- Piao, J.; Chen, L.; Quan, T.; Li, L.; Quan, C.; Piao, Y.; Jin, T.; Lin, Z. Superior efficacy of co-treatment with the dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A against NSCLC. Oncotarget 2016, 7, 60169–60180. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Edell, K.A.; Yevtushenko, M.A.; Rothschild, D.E.; Rogers, A.N.; Benz, C.C. mTORC1/C2 and pan-HDAC inhibitors synergistically impair breast cancer growth by convergent AKT and polysome inhibiting mechanisms. Breast Cancer Res. Treat. 2014, 144, 287–298. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Tsaur, I.; Chun, F.K.-H.; Bereiter-Hahn, J.; Blaheta, R.A. Influence of the HDAC Inhibitor Valproic Acid on the Growth and Proliferation of Temsirolimus-Resistant Prostate Cancer Cells In Vitro. Cancers 2019, 11, 566. https://doi.org/10.3390/cancers11040566
Makarević J, Rutz J, Juengel E, Maxeiner S, Tsaur I, Chun FK-H, Bereiter-Hahn J, Blaheta RA. Influence of the HDAC Inhibitor Valproic Acid on the Growth and Proliferation of Temsirolimus-Resistant Prostate Cancer Cells In Vitro. Cancers. 2019; 11(4):566. https://doi.org/10.3390/cancers11040566
Chicago/Turabian StyleMakarević, Jasmina, Jochen Rutz, Eva Juengel, Sebastian Maxeiner, Igor Tsaur, Felix K.-H. Chun, Jürgen Bereiter-Hahn, and Roman A. Blaheta. 2019. "Influence of the HDAC Inhibitor Valproic Acid on the Growth and Proliferation of Temsirolimus-Resistant Prostate Cancer Cells In Vitro" Cancers 11, no. 4: 566. https://doi.org/10.3390/cancers11040566
APA StyleMakarević, J., Rutz, J., Juengel, E., Maxeiner, S., Tsaur, I., Chun, F. K.-H., Bereiter-Hahn, J., & Blaheta, R. A. (2019). Influence of the HDAC Inhibitor Valproic Acid on the Growth and Proliferation of Temsirolimus-Resistant Prostate Cancer Cells In Vitro. Cancers, 11(4), 566. https://doi.org/10.3390/cancers11040566