TRPC3 Regulates the Proliferation and Apoptosis Resistance of Triple Negative Breast Cancer Cells through the TRPC3/RASA4/MAPK Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
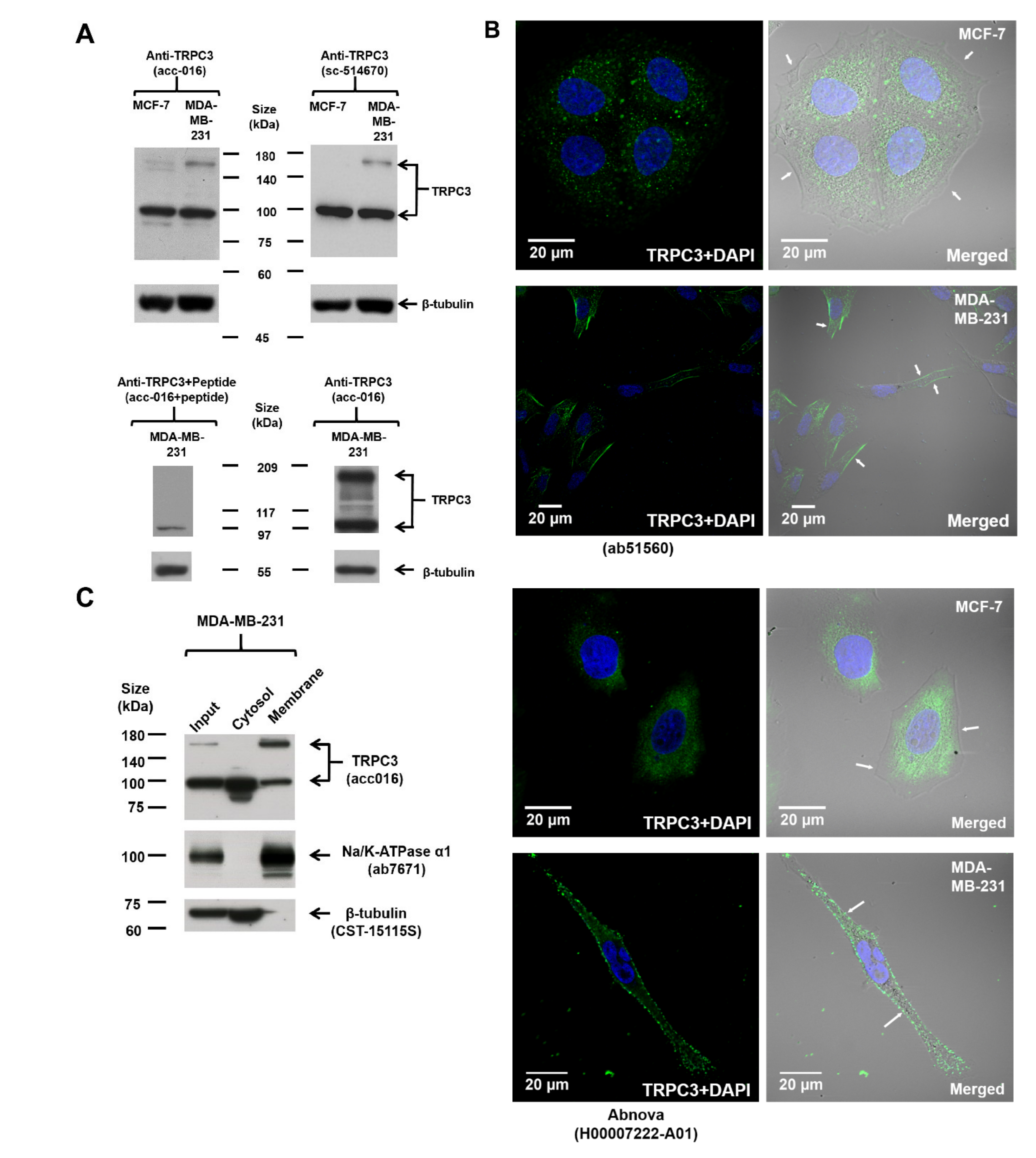
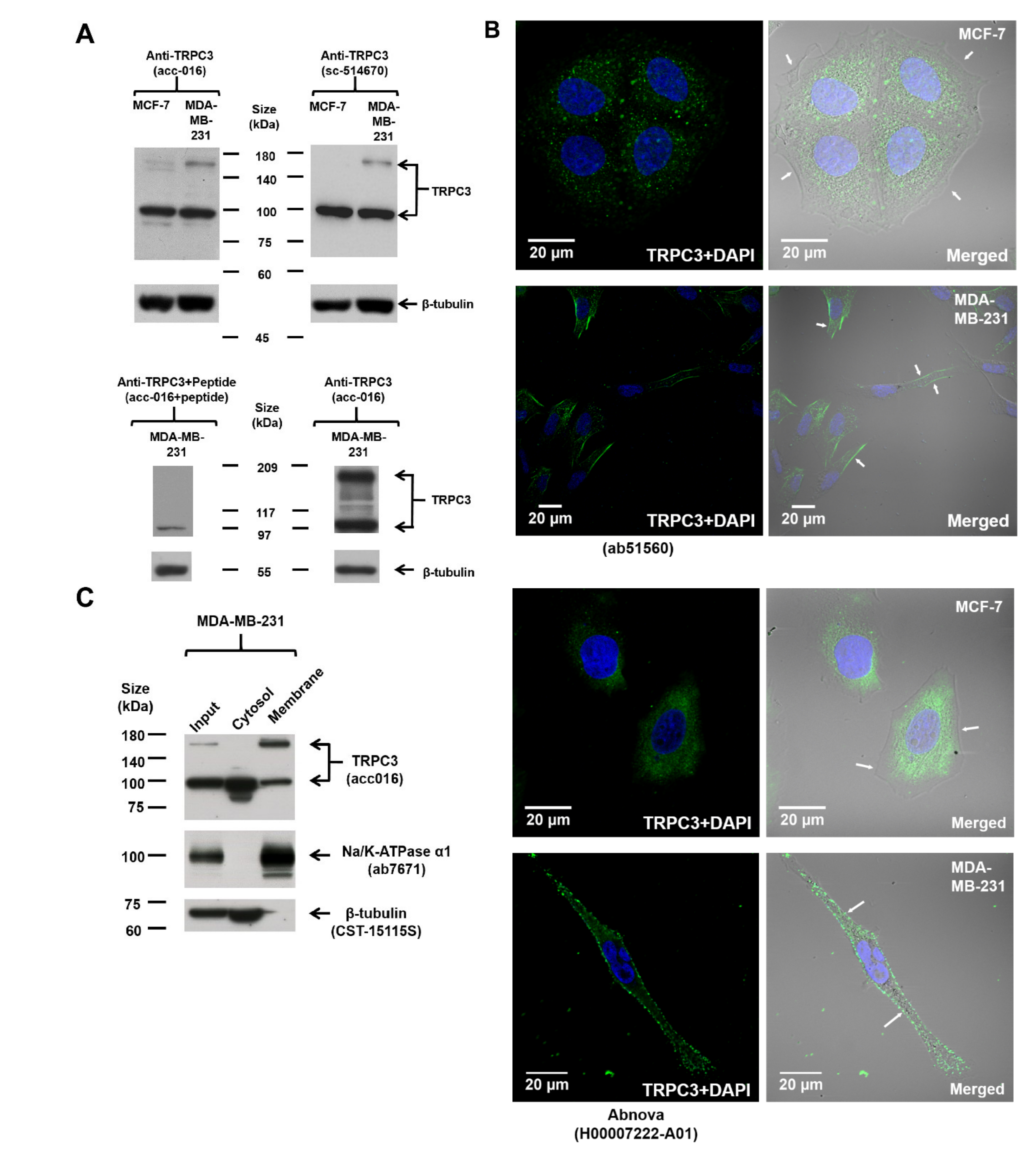
2.1. Upregulation of TRPC3 on the Plasma Membrane of Triple-Negative Breast Cancer (TNBC) Cells MDA-MB-231
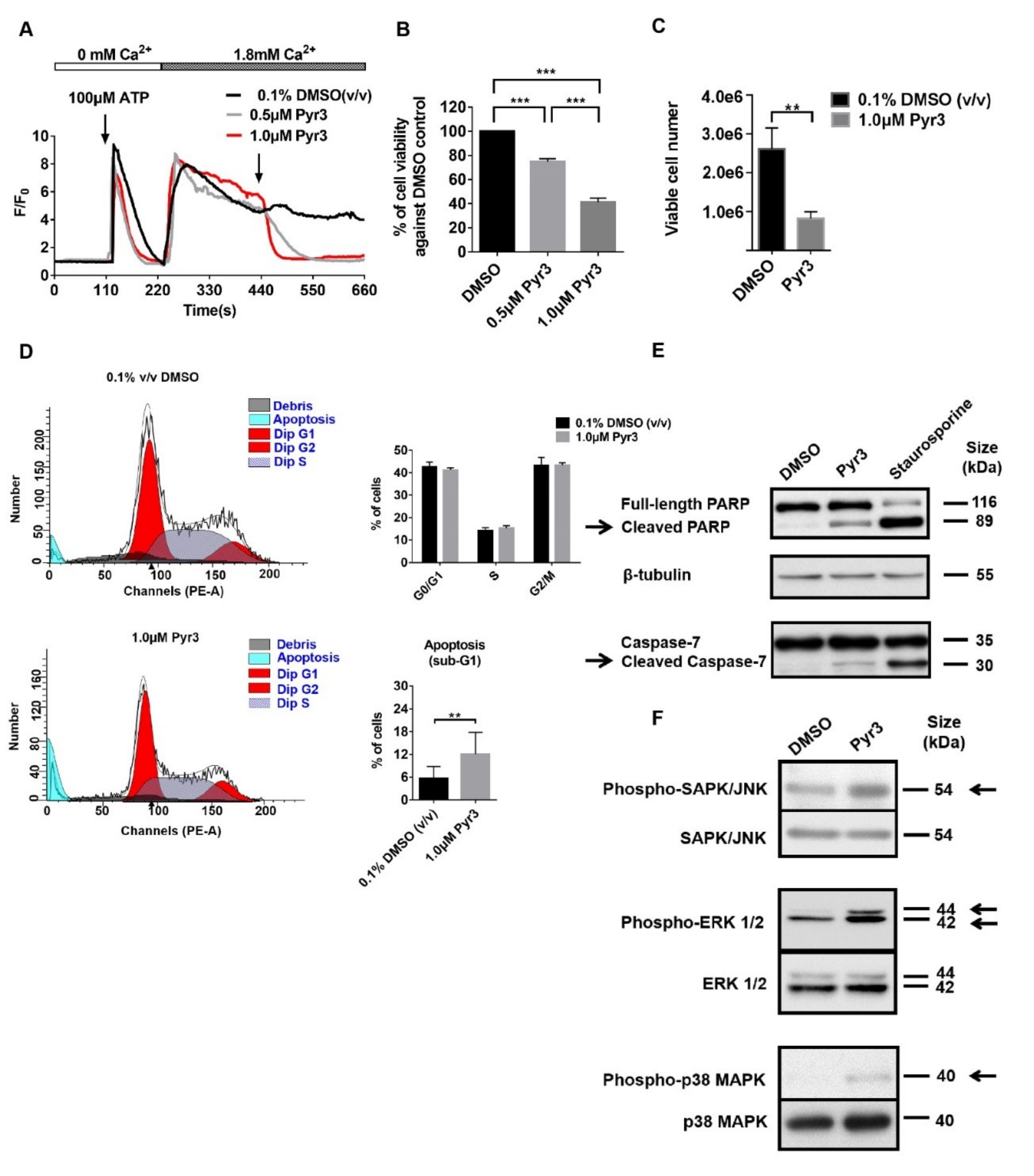
2.2. TRPC3 Regulated Calcium Influx, Cell Proliferation and Apoptosis of MDA-MB-231
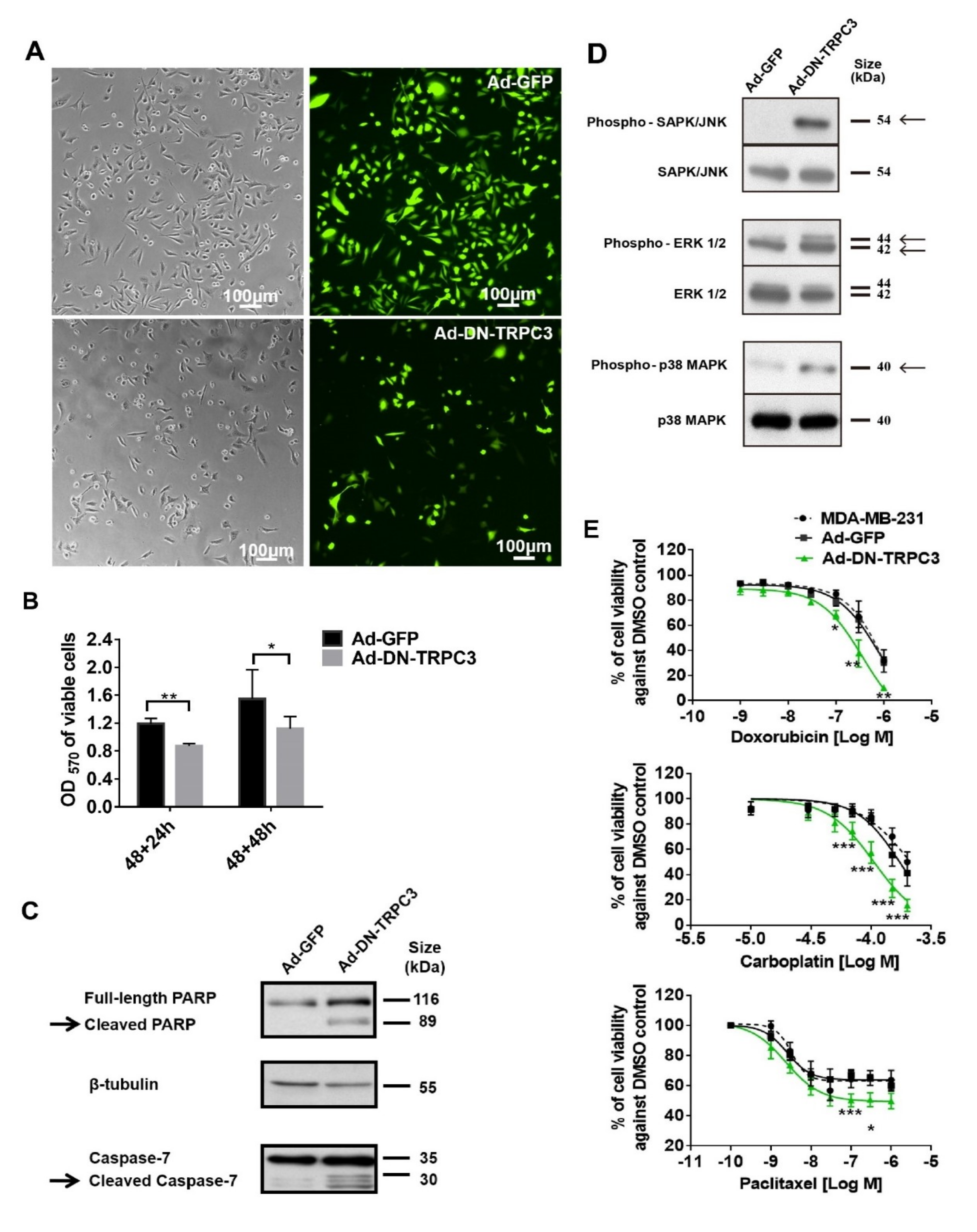
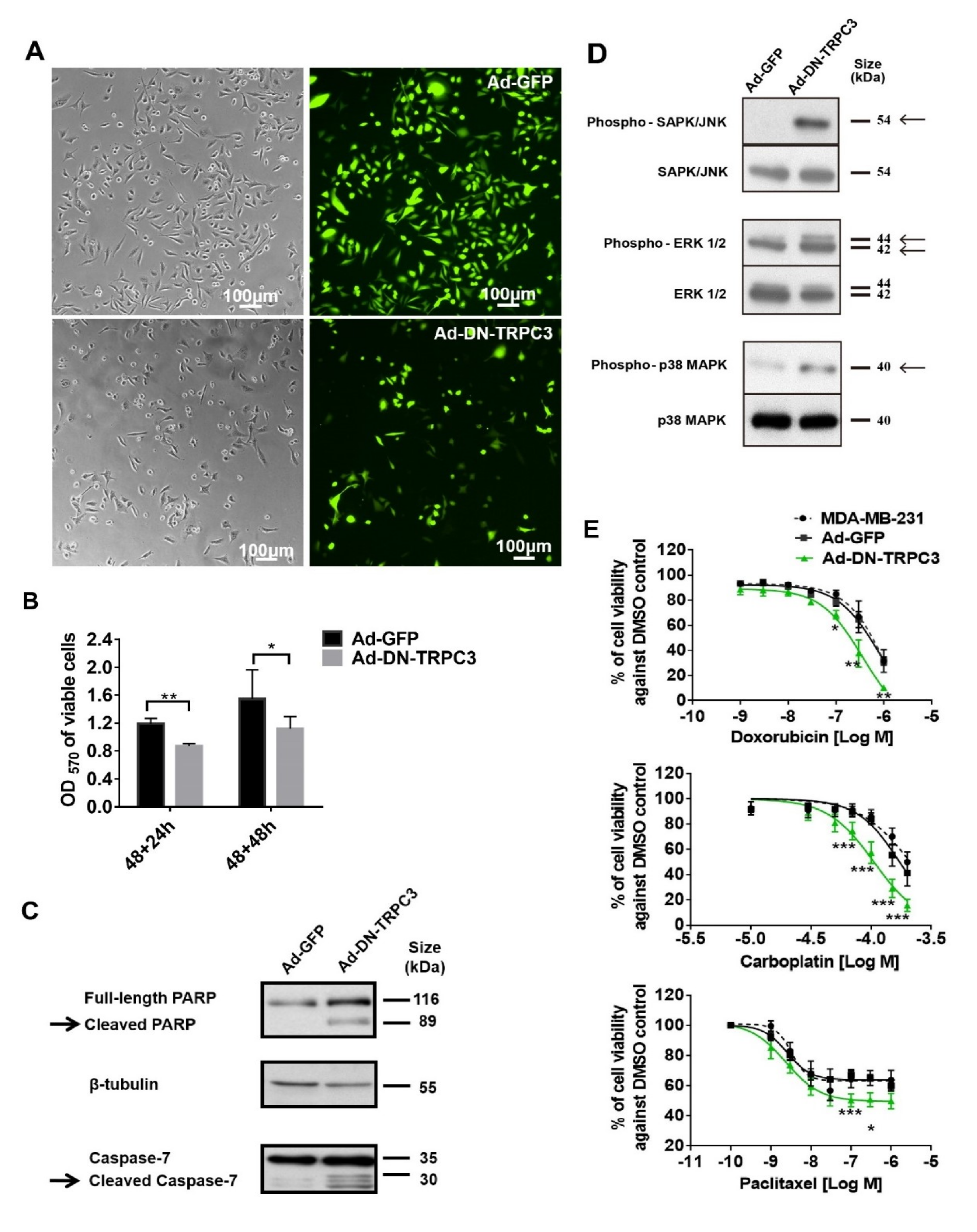
2.3. Dominant Negative (DN) of TRPC3 Attenuated Cell Proliferation, Induced Cell Apoptosis and Sensitized Cell Death to Chemotherapeutic Agents in MDA-MB-231
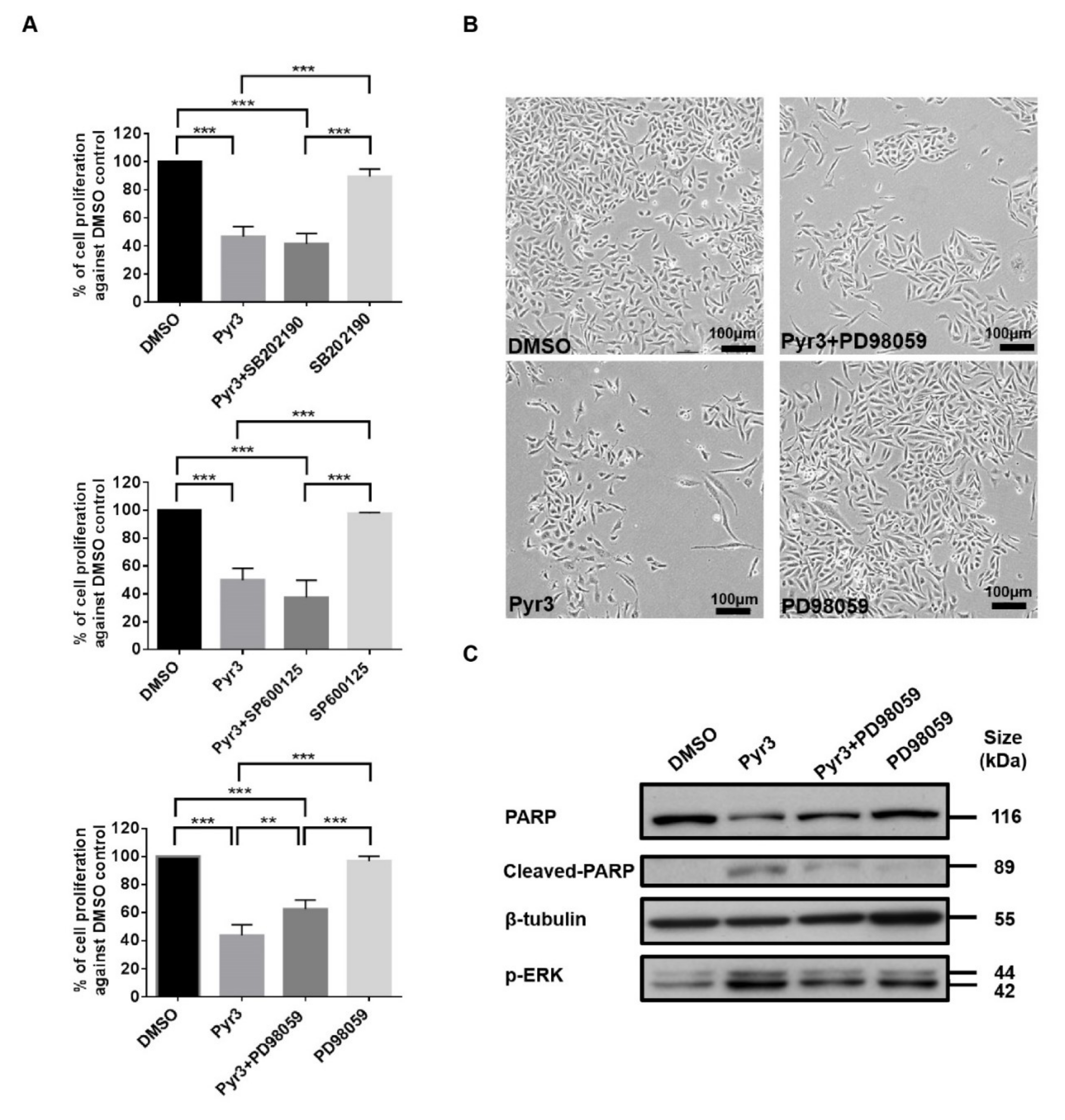
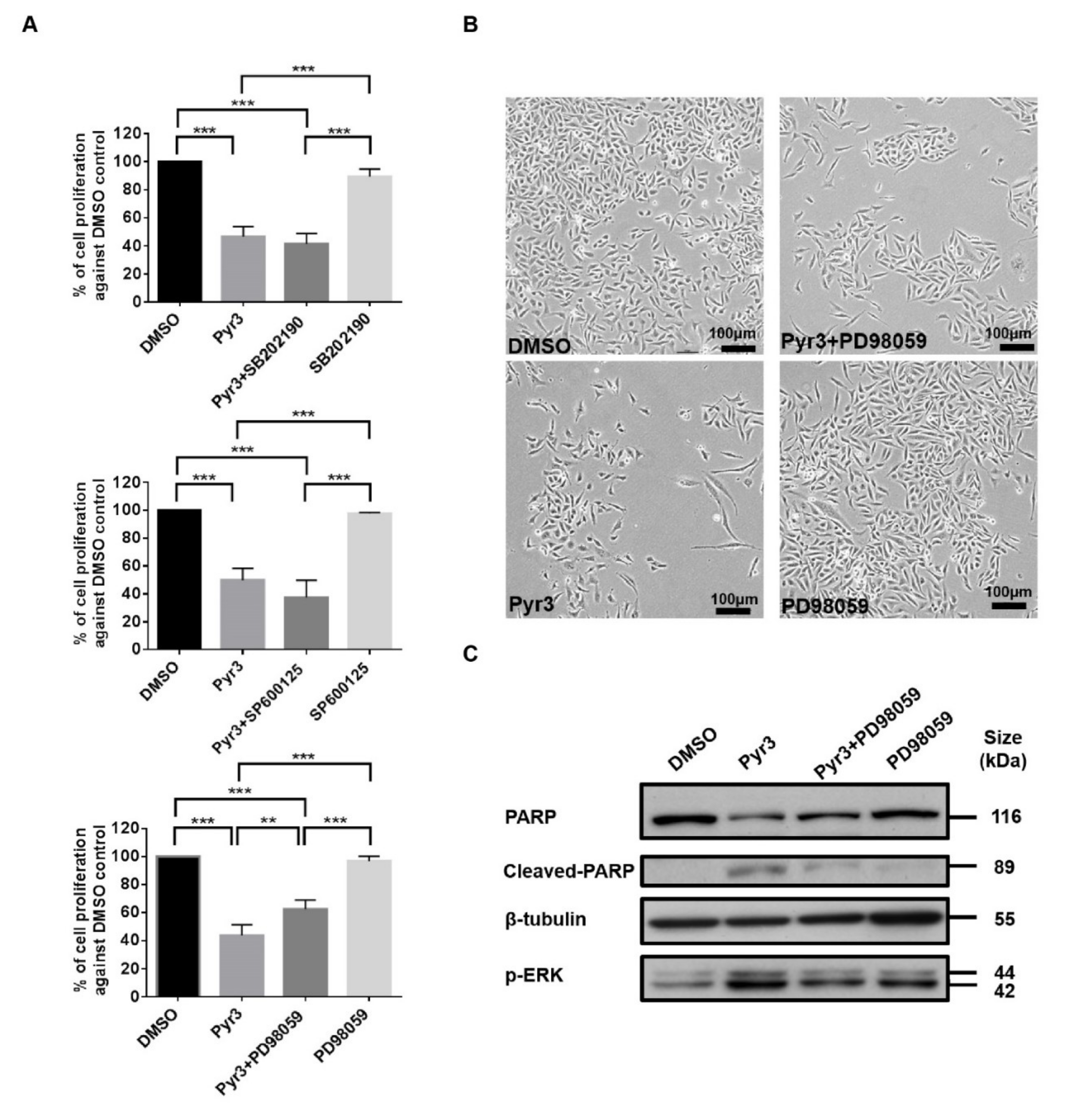
2.4. TRPC3 Blockade Induced Apoptosis in MDA-MB-231 Cells Activation of ERK 1/2
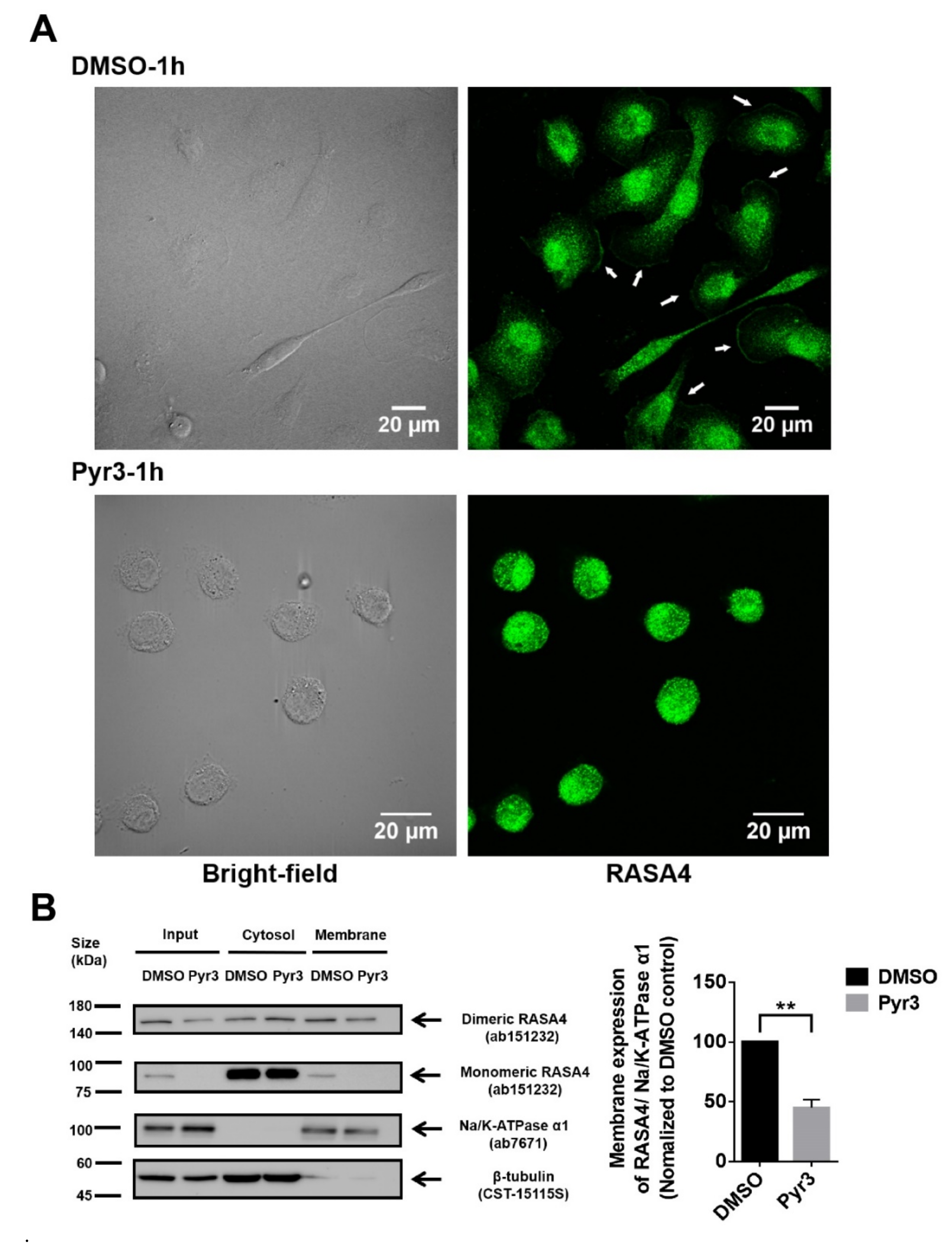
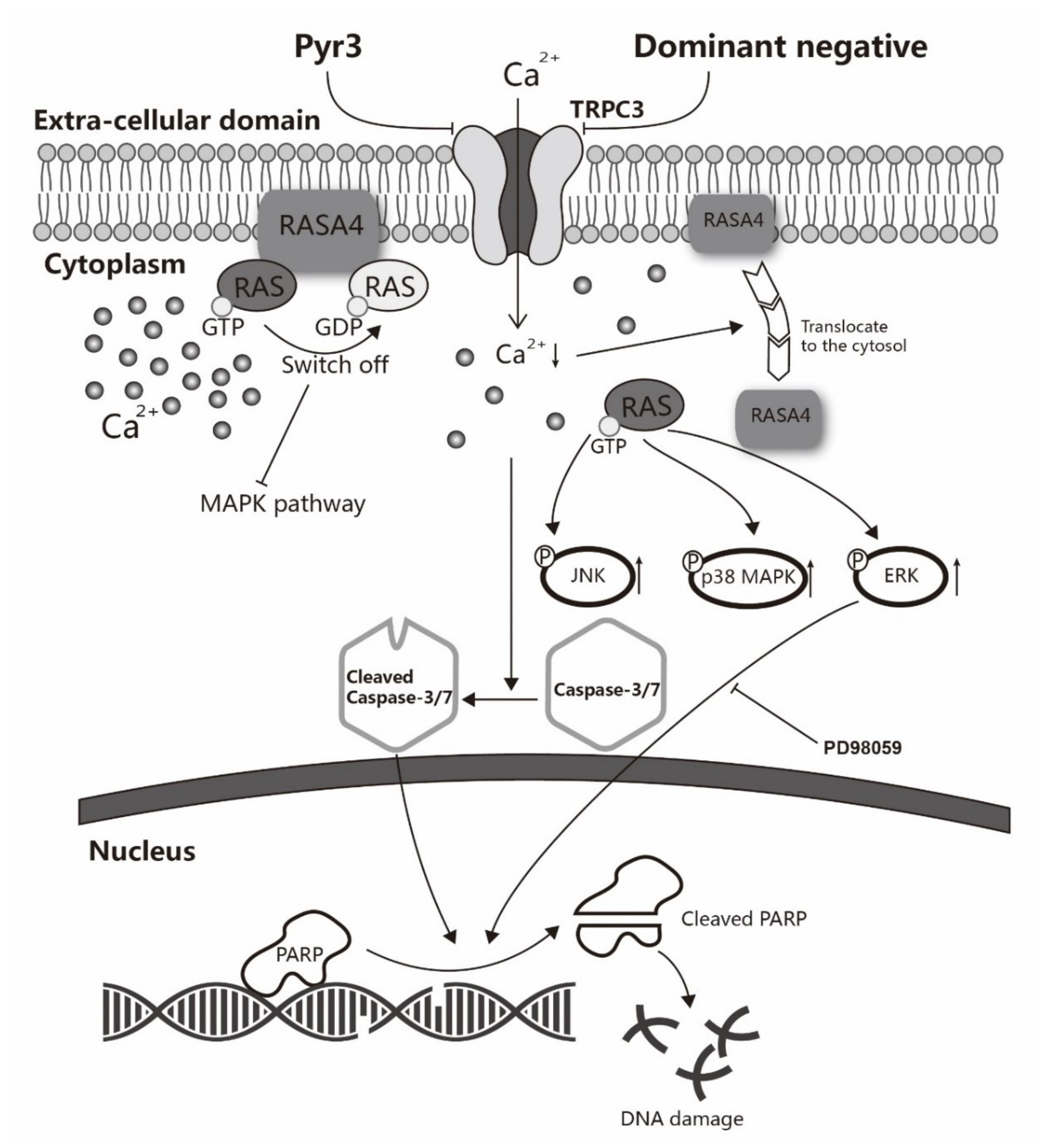
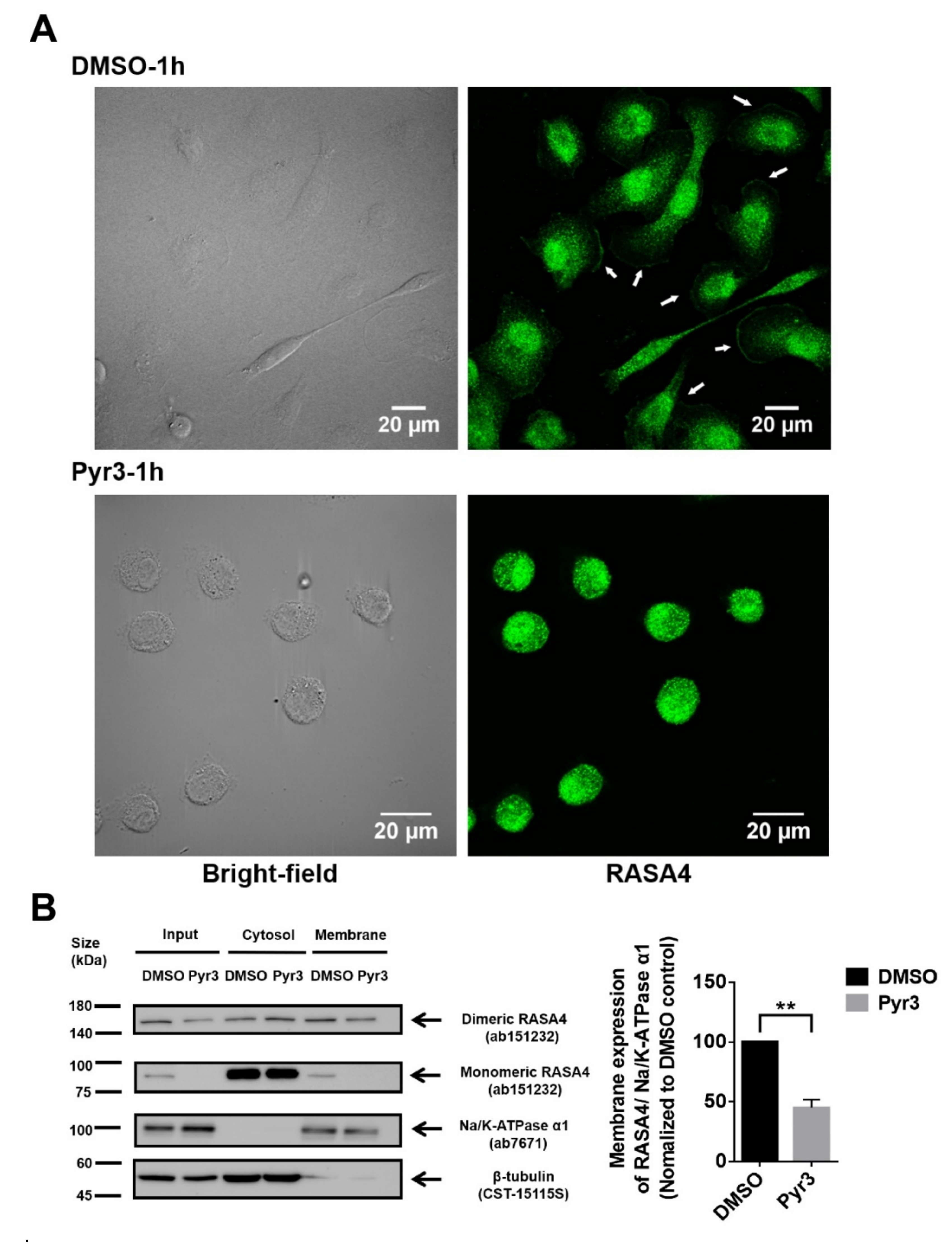
2.5. Involvement of RASA4 in TRPC3-Mediated Calcium Signaling Transduction
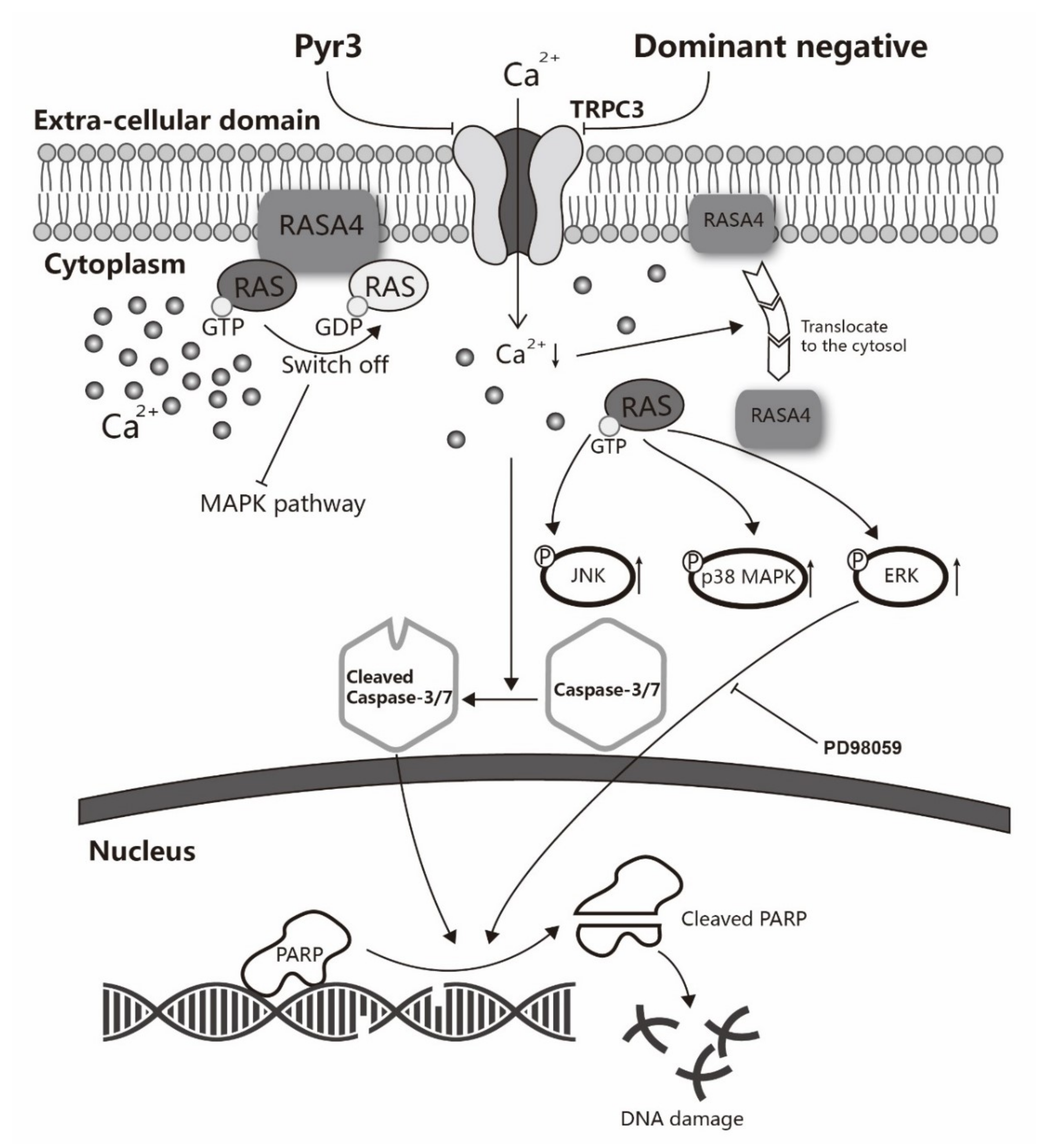
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatment Regimen
4.3. Western Blot
4.4. Immunocytochemistry
4.5. Subcellular Fractionation Followed by Western Blot
4.6. Confocal Ca2+ Imaging
4.7. Proliferation Assay
4.8. Propidium Iodide (PI) Staining Followed by Flow Cytometry for Cell Cycle Analysis
4.9. Fluorescence Imaging
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 10, 3786–3788. [Google Scholar] [CrossRef]
- Sims, A.H.; Howell, A.; Howell, S.J.; Clarke, R.B. Origins of breast cancer subtypes and therapeutic implications. Nat. Clin. Pract. Oncol. 2007, 4, 516–525. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Debatin, K.M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar] [CrossRef]
- Munshi, A.; Ramesh, R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjuhates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Azimi, I.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef]
- Gkika, D.; Prevarskaya, N. Molecular mechanisms of TRP regulation in tumor growth and metastasis. Biochim. Biophys. Acta 2009, 1793, 953–958. [Google Scholar] [CrossRef]
- Ouadid-Ahidouch, H.; Dhennin-Duthille, I.; Gautier, M.; Sevestre, H.; Ahidouch, A. TRP channels: Diagnostic markers and therapeutic targets for breast cancer? Trends Mol. Med. 2013, 19, 117–124. [Google Scholar] [CrossRef]
- Aydar, E.; Yeo, S.; Djamgoz, M.; Palmer, C. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: A potential target for breast cancer diagnosis and therapy. Cancer Cell Int. 2009, 9, 23–35. [Google Scholar] [CrossRef]
- Yang, S.L.; Cao, Q.; Zhou, K.C.; Feng, Y.J.; Wang, Y.Z. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 2009, 28, 1320–1328. [Google Scholar] [CrossRef]
- Lichtenegger, M.; Stockner, T.; Poteser, M.; Schleifer, H.; Platzer, D.; Romanin, C.; Groschner, K. A novel homology model of TRPC3 reveals allosteric coupling between gate and selectivity filter. Cell Calcium 2013, 54, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Lintschinger, B.; Balzer-Geldsetzer, M.; Baskaran, T.; Graier, W.F.; Romanin, C.; Zhu, M.X.; Groschner, K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J. Biol. Chem. 2000, 275, 27799–27805. [Google Scholar] [CrossRef] [PubMed]
- Zagranichnaya, T.K.; Wu, X.; Villereal, M.L. Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in HEK-293 cells. J. Biol. Chem. 2005, 280, 29559–29569. [Google Scholar] [CrossRef]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y.; Yoshida, T.; Wakamori, M.; Mori, E.; Numata, T.; et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Wong, C.K.; Suen, C.H.; Wang, J.; Long, C.; Sauer, H.; Yao, X.; Tsang, S.Y. TRPC3 regulates the automaticity of embryonic stem cell-derived cardiomyocytes. Int. J. Cardiol. 2016, 203, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Yung, L.H.; Wong, W.T.; Liu, J.; Leung, F.P.; Liu, L.; Chen, Y.; Kong, S.K.; Kwan, K.M.; Ng, S.M.; et al. Bone morphogenic protein-4 induces endothelial cell apoptosis through oxidative stress-dependent p38 MAPK and JNK pathway. J. Mol. Cell. Cardiol. 2012, 52, 237–244. [Google Scholar] [CrossRef]
- Cullen, P.J.; Lockyer, P.J. Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 339–348. [Google Scholar] [CrossRef]
- Kupzig, S.; Walker, S.A.; Cullen, P.J. The frequencies of calcium oscillations are optimized for efficient calcium-mediated activation of Ras and the ERK/MAPK cascade. Proc. Natl. Acad. Sci. USA 2005, 102, 7577–7582. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, J.; Dzhagalov, I.; He, Y.W. An essential function for the calcium-promoted Ras inactivator in Fcgamma receptor-mediated phagocytosis. Nat. Immunol. 2005, 6, 911–919. [Google Scholar] [CrossRef]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 channels are required for proliferation, migration and invasion of breast cancer cell lines by modulation of Orai1 and Orai3 surface exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.T.D.S.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef]
- Jiang, H.N.; Zeng, B.; Zhang, Y.; Daskoulidou, N.; Fan, H.; Qu, J.M.; Xu, S.Z. Involvement of TRPC channels in lung cancer cell differentiation and the correlation analysis in human non-small cell lung cancer. PLoS ONE 2013, 8, e67637. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Montagna, M.K.; Holmberg, J.C.; Craveiro, V.; Brown, D.; Mor, G. Targeting the mitochondia activates two independent cell death pathways in ovarian cancer stem cells. Mol. Cancer Ther. 2011, 10, 1385–1393. [Google Scholar] [CrossRef]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef]
- Tao, X.; Zhao, N.; Jin, H.; Zhang, Z.; Liu, Y.; Wu, J.; Bast, R.C.J.; Yu, Y.; Feng, Y. FSH enhances the proliferation of ovarian cancer cells by activating transient receptor potential channel C3. Endocr. Relat. Cancer 2013, 20, 415–429. [Google Scholar] [CrossRef]
- Saito, H.; Minamiya, Y.; Watanabe, H.; Takahashi, N.; Ito, M.; Toda, H.; Konno, H.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of the transient receptor potential channel c3 correlates with a favorable prognosis in patients with adenocarcinoma of the lung. Ann. Surg. Oncol. 2011, 18, 3377–3383. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.; Ganzinelli, M.; Andreis, D.; Bertoni, R.; Giardini, R.; Fox, S.B.; Broggini, M.; Bottini, A.; Zanoni, V.; Bazzola, L.; et al. Triple negative breast cancers have a reduced expression of DNA repair genes. PLoS ONE 2013, 8, e66243. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Kitazumi, I.; Tsukahara, M. Regulation of DNA fragmentation: The role of caspases and phosphorylation. FEBS J. 2011, 278, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert. Opin. Ther. Targets 2012, 216, 103–119. [Google Scholar] [CrossRef]
- Azoitei, N.; Hoffmann, C.M.; Ellegast, J.M.; Ball, C.R.; Obermayer, K.; Gößele, U.; Koch, B.; Faber, K.; Genze, F.; Schrader, M.; et al. Targeting of KRAS mutant tumors by HSP90 inhibitors involves degradation of STK33. J. Exp. Med. 2012, 209, 697–711. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, S. ERK1/2 MAP Kinases in cell survival and apoptosis. IUBMB Life 2006, 158, 621–631. [Google Scholar] [CrossRef]
- Martin, M.C.; Allan, L.A.; Mancini, E.J.; Clarke, P.R. The docking interaction of caspase-9 with ERK2 provides a mechanism for the selective inhibitory phosphorylation of caspase-9 at threonine 125. J. Biol. Chem. 2008, 283, 3854–3865. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Chouhan, S.; Singh, S.; Chhipa, R.R.; Ajay, A.K.; Bhat, M.K. Constitutively activated ERK sensitizes cancer cells to doxorubicin: Involvement of p53-EGFR-ERK pathway. J. Biosci. 2017, 42, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Upadhyay, A.K.; Ajay, A.K.; Bhat, M.K. p53 regultes ERK activation in carboplatin induced apoptosis in cervical carcinoma: A novel target of p53 in apoptosis. FEBS Lett. 2007, 581, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Lim, W.; Ham, J.; Bazer, F.W.; Song, G. Carvarcol induces mitochondria-mediated apoptosis via disruption of calcium homemtasis in human choriocarcinoma cells. J. Cell. Physiol. 2019, 234, 1803–1815. [Google Scholar] [CrossRef]
- Abdoul-Azize, S.; Buquet, C.; Li, H.; Picquenot, J.M.; Vannier, J.P. Integration of Ca2+ signaling regulates the breast tumor cell response to simvastatin and doxorubicin. Oncogenge 2018, 37, 4979–4993. [Google Scholar] [CrossRef]
- Monet, M.; Lehen’kyi, V.; Gackiere, F.; Firlej, V.; Vandenberghe, M.; Roudbaraki, M.; Gkika, D.; Pourtier, A.; Bidaux, G.; Slomianny, C.; et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010, 70, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Raphaël, M.; Lehen’kyi, V.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Vanden, A.F.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer cells co-opt the neuronal redoxsensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell 2018, 33, 985–1003. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Bao, Y.; Li, Z.; Li, J.; Gong, M.; Lam, S.; Wang, J.; Marzese, D.M.; Donovan, N.; Tan, E.Y.; et al. RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J. Clin. Investig. 2014, 124, 5291–5304. [Google Scholar] [CrossRef]
- Lo, I.C.; Chan, H.C.; Qi, Z.; Ng, K.L.; So, C.; Tsang, S.Y. TRPV3 channel negatively regulates cell cycle progression and safeguards the pluripotency of embryonic stem cells. J. Cell. Physiol. 2016, 231, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Groschner, K.; Hingel, S.; Lintschinger, B.; Balzer, M.; Romanin, C.; Zhu, X.; Schreibmayer, W. Trp proteins form store-operated cation channels in human vascular endothelial cells. FEBS Lett. 1998, 437, 101–106. [Google Scholar] [CrossRef]
- Xu, Y.; So, C.; Lam, H.M.; Fung, M.C.; Tsang, S.Y. Apoptosis reversal promotes cancer stem cell-like cell formation. Neoplasia 2018, 20, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Tiapko, O.; Groschner, K. TRPC3 as a target of novel therapeutic interventions. Cells 2018, 7, 83. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Qi, Y.-X.; Qi, Z.; Tsang, S.-Y. TRPC3 Regulates the Proliferation and Apoptosis Resistance of Triple Negative Breast Cancer Cells through the TRPC3/RASA4/MAPK Pathway. Cancers 2019, 11, 558. https://doi.org/10.3390/cancers11040558
Wang Y, Qi Y-X, Qi Z, Tsang S-Y. TRPC3 Regulates the Proliferation and Apoptosis Resistance of Triple Negative Breast Cancer Cells through the TRPC3/RASA4/MAPK Pathway. Cancers. 2019; 11(4):558. https://doi.org/10.3390/cancers11040558
Chicago/Turabian StyleWang, Yan, Yan-Xiang Qi, Zenghua Qi, and Suk-Ying Tsang. 2019. "TRPC3 Regulates the Proliferation and Apoptosis Resistance of Triple Negative Breast Cancer Cells through the TRPC3/RASA4/MAPK Pathway" Cancers 11, no. 4: 558. https://doi.org/10.3390/cancers11040558
APA StyleWang, Y., Qi, Y.-X., Qi, Z., & Tsang, S.-Y. (2019). TRPC3 Regulates the Proliferation and Apoptosis Resistance of Triple Negative Breast Cancer Cells through the TRPC3/RASA4/MAPK Pathway. Cancers, 11(4), 558. https://doi.org/10.3390/cancers11040558