Germline Missense Variants in BRCA1: New Trends and Challenges for Clinical Annotation


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
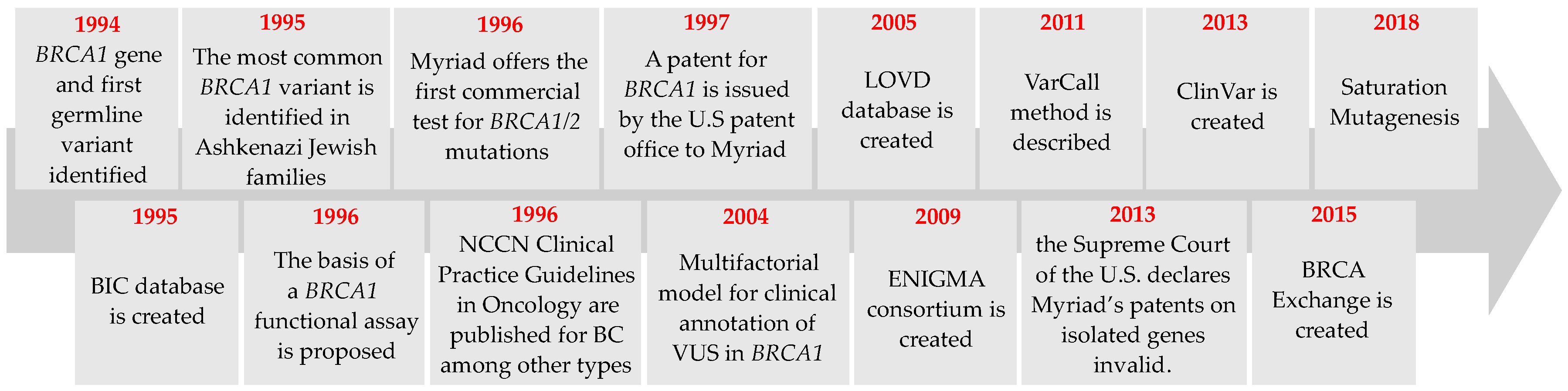
2. BRCA1 Variant Detection: From First Variants to New Trends
2.1. Detection of the First Germline Variants in BRCA1
2.2. Commercial Testing
2.3. Genetic Testing Developments
2.4. The Spectrum of BRCA1 Variants across the World
3. BRCA1 Variant Annotation and Classification
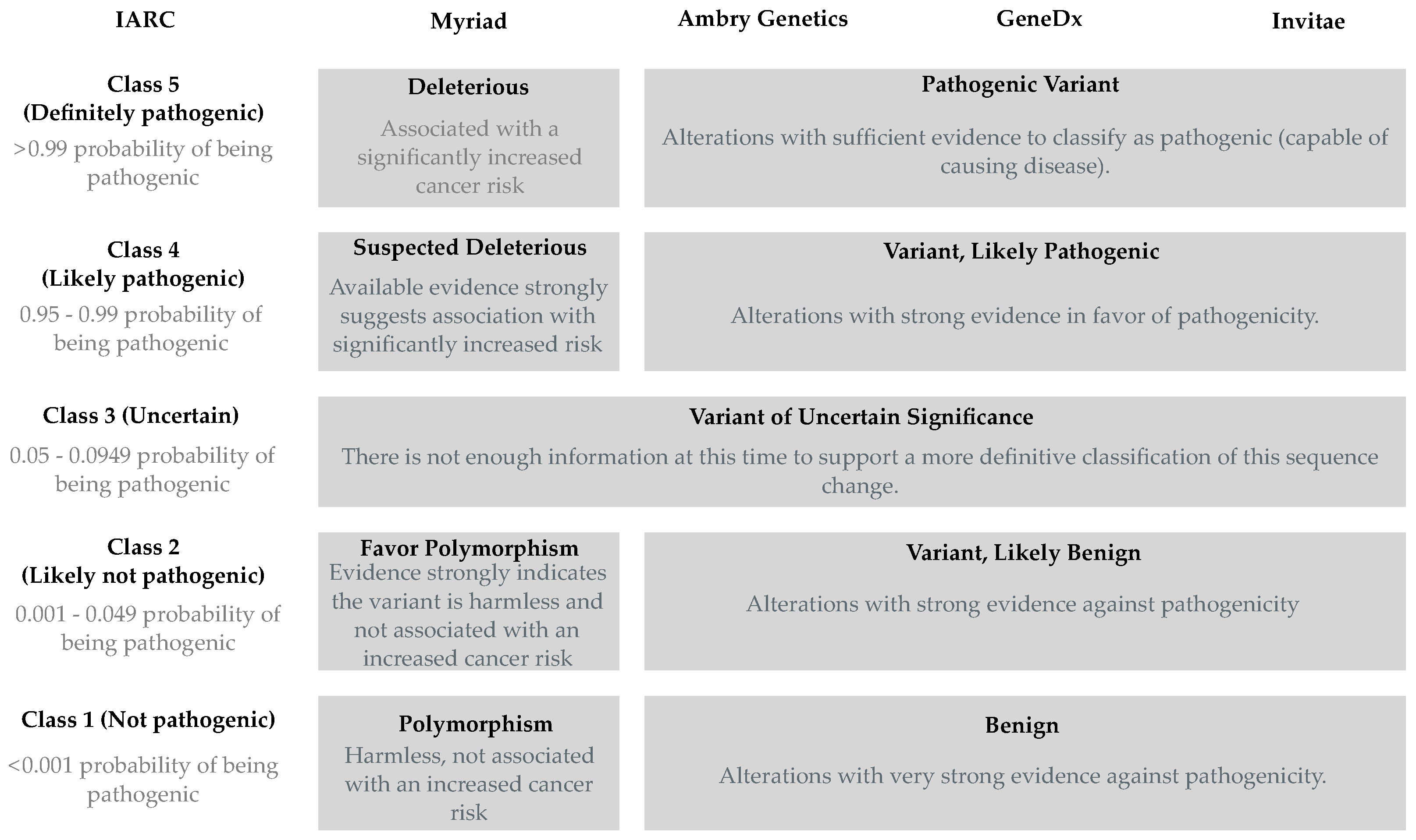
3.1. Definitions and Limitations
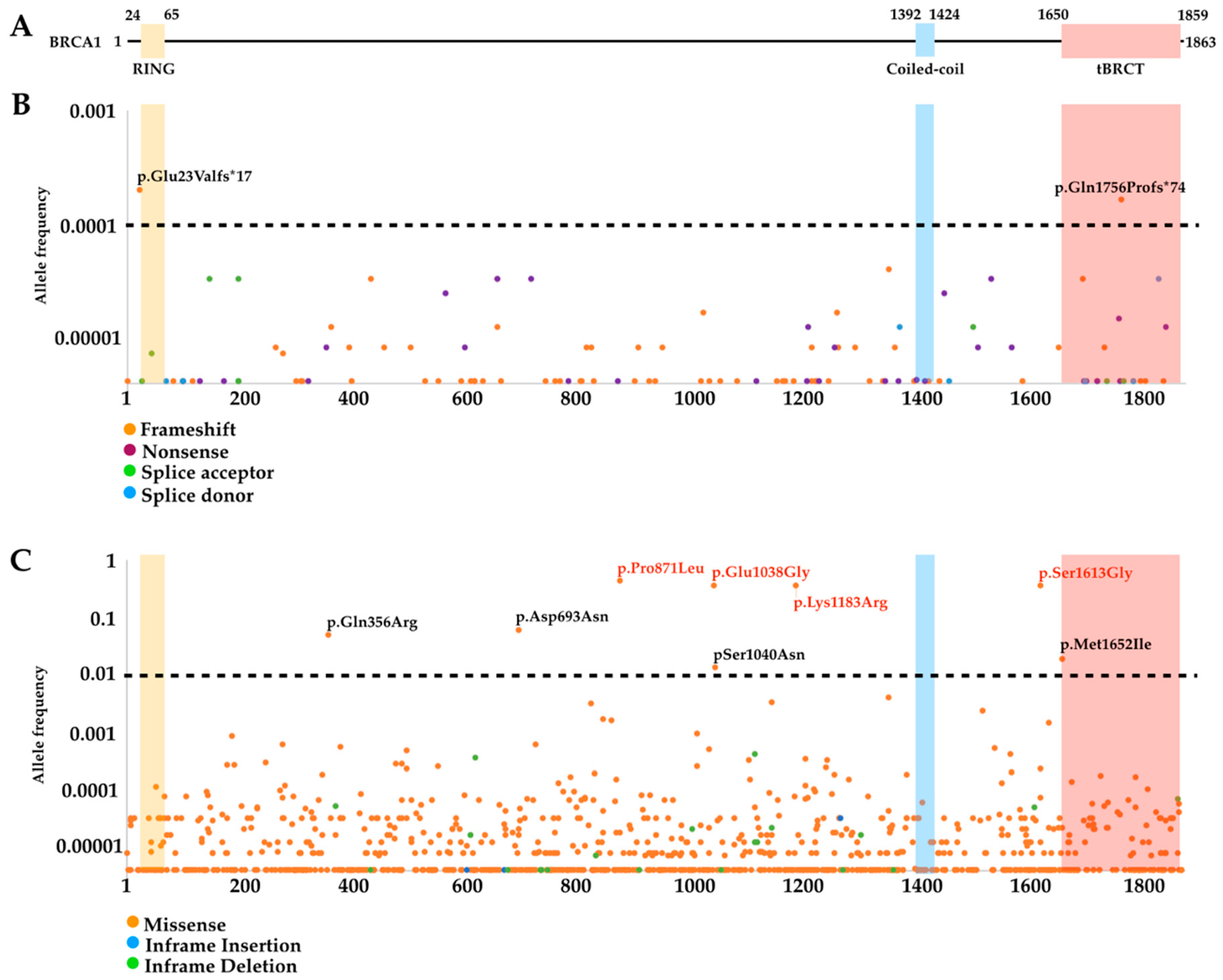
3.2. Mutational Landscape
3.3. Variants of Uncertain Significance (VUS)
3.4. Functional Assays
4. BRCA1 Variant Databases and Information Dissemination
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Brinton, L.A.; Gaudet, M.M.; Gierach, G.L. Breast cancer. In Cancer Epidemiology and Prevention, 4th ed.; Thun, M., Linet, M.S., Cerhan, J.R., Haiman, C.A., Schottenfeld, D., Eds.; Oxford University Press: New York, NY, USA, 2018. [Google Scholar]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA: A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Olopade, O.I. Breast cancer risk associated with brca1 and brca2 in diverse populations. Nat. Rev. Cancer 2007, 7, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Alsop, K.; Fereday, S.; Meldrum, C.; de Fazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. Brca mutation frequency and patterns of treatment response in brca mutation-positive women with ovarian cancer: A report from the australian ovarian cancer study group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for brca1 and brca2 mutation carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Network, N.C.C. Genetic/Familial High-Risk Assessment: Breast and Ovarian (Version 3.2019). Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf (accessed on 19 October 2018).
- Gadzicki, D.; Evans, D.G.; Harris, H.; Julian-Reynier, C.; Nippert, I.; Schmidtke, J.; Tibben, A.; van Asperen, C.J.; Schlegelberger, B. Genetic testing for familial/hereditary breast cancer-comparison of guidelines and recommendations from the uk, france, the netherlands and germany. J. Community Genet. 2011, 2, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Bonaiti-Pellie, C.; Andrieu, N.; Arveux, P.; Bonadona, V.; Buecher, B.; Delpech, M.; Jolly, D.; Julian-Reynier, C.; Luporsi, E.; Nogues, C.; et al. Cancer genetics: Estimation of the needs of the population in france for the next ten years. Bull Cancer 2009, 96, 875–900. [Google Scholar]
- Chavarri-Guerra, Y.; Blazer, K.R.; Weitzel, J.N. Genetic cancer risk assessment for breast cancer in latin america. Rev. Investig. Clin. 2017, 69, 94–102. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA challenge: BRCA exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Futreal, P.A.; Liu, Q.; Shattuck-Eidens, D.; Cochran, C.; Harshman, K.; Tavtigian, S.; Bennett, L.M.; Haugen-Strano, A.; Swensen, J.; Miki, Y. BRCA1 mutations in primary breast and ovarian carcinomas. Science 1994, 266, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Behbakht, K.; McGovern, P.E.; Chiu, H.C.; Couch, F.J.; Weber, B.L.; Friedman, L.S.; King, M.C.; Furusato, M.; LiVolsi, V.A. Mutation analysis of the BRCA1 gene in ovarian cancers. Cancer Res. 1995, 55, 2998–3002. [Google Scholar]
- Merajver, S.D.; Pham, T.M.; Caduff, R.F.; Chen, M.; Poy, E.L.; Cooney, K.A.; Weber, B.L.; Collins, F.S.; Johnston, C.; Frank, T.S. Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat. Genet. 1995, 9, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Hosking, L.; Trowsdale, J.; Nicolai, H.; Solomon, E.; Foulkes, W.; Stamp, G.; Signer, E.; Jeffreys, A. A somatic brca1 mutation in an ovarian tumour. Nat. Genet. 1995, 9, 343–344. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Andermann, A.; Narod, S.A. Genetic counselling for familial breast and ovarian cancer in ontario. J. Med Genet. 2002, 39, 695–696. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Supreme Court of the United States. Association for molecular pathology v. Myriad genetics, Inc; Supreme Court of the United States: Washington, DC, USA, 2013; Volume 569 U. S. 576.
- Kesselheim, A.S.; Cook-Deegan, R.M.; Winickoff, D.E.; Mello, M.M. Gene patenting—The supreme court finally speaks. N. Engl. J. Med. 2013, 369, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Hagen, A.I.; Apold, J.; Maehle, L.; Clark, N.; Fiane, B.; Lovslett, K.; Hovig, E.; Vabo, A. Genetic epidemiology of BRCA mutations—Family history detects less than 50% of the mutation carriers. Eur. J. Cancer 2007, 43, 1713–1717. [Google Scholar] [CrossRef]
- Johannsson, O.; Ostermeyer, E.A.; Hakansson, S.; Friedman, L.S.; Johansson, U.; Sellberg, G.; Brondum-Nielsen, K.; Sele, V.; Olsson, H.; King, M.C.; et al. Founding BRCA1 mutations in hereditary breast and ovarian cancer in southern Sweden. Am. J. Hum. Genet. 1996, 58, 441–450. [Google Scholar]
- Neuhausen, S.L.; Godwin, A.K.; Gershoni-Baruch, R.; Schubert, E.; Garber, J.; Stoppa-Lyonnet, D.; Olah, E.; Csokay, B.; Serova, O.; Lalloo, F.; et al. Haplotype and phenotype analysis of nine recurrent BRCA2 mutations in 111 families: Results of an international study. Am. J. Hum. Genet. 1998, 62, 1381–1388. [Google Scholar] [CrossRef]
- Schubert, E.L.; Lee, M.K.; Mefford, H.C.; Argonza, R.H.; Morrow, J.E.; Hull, J.; Dann, J.L.; King, M.C. BRCA2 in american families with four or more cases of breast or ovarian cancer: Recurrent and novel mutations, variable expression, penetrance, and the possibility of families whose cancer is not attributable to BRCA1 or BRCA2. Am. J. Hum. Genet. 1997, 60, 1031–1040. [Google Scholar]
- U.S. Food and Drug Administration. Genetic Health Risk Assessment System. In Code of Federal Regulations; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2018; Volume 21CFR866.5950. [Google Scholar]
- Gabai-Kapara, E.; Lahad, A.; Kaufman, B.; Friedman, E.; Segev, S.; Renbaum, P.; Beeri, R.; Gal, M.; Grinshpun-Cohen, J.; Djemal, K.; et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc. Natl. Acad. Sci. USA 2014, 111, 14205–14210. [Google Scholar] [CrossRef]
- Frank, T.S.; Deffenbaugh, A.M.; Reid, J.E.; Hulick, M.; Ward, B.E.; Lingenfelter, B.; Gumpper, K.L.; Scholl, T.; Tavtigian, S.V.; Pruss, D.R.; et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: Analysis of 10,000 individuals. J. Clin. Oncol. 2002, 20, 1480–1490. [Google Scholar] [CrossRef]
- Frank, T.S.; Manley, S.A.; Olopade, O.I.; Cummings, S.; Garber, J.E.; Bernhardt, B.; Antman, K.; Russo, D.; Wood, M.E.; Mullineau, L.; et al. Sequence analysis of BRCA1 and BRCA2: Correlation of mutations with family history and ovarian cancer risk. J. Clin. Oncol. 1998, 16, 2417–2425. [Google Scholar] [CrossRef]
- Serova-Sinilnikova, O.M.; Boutrand, L.; Stoppa-Lyonnet, D.; Bressac-de-Paillerets, B.; Dubois, V.; Lasset, C.; Janin, N.; Bignon, Y.J.; Longy, M.; Maugard, C.; et al. BRCA2 mutations in hereditary breast and ovarian cancer in france. Am. J. Hum. Genet. 1997, 60, 1236–1239. [Google Scholar]
- Lee, K.; Seifert, B.A.; Shimelis, H.; Ghosh, R.; Crowley, S.B.; Carter, N.J.; Doonanco, K.; Foreman, A.K.; Ritter, D.I.; Jimenez, S.; et al. Clinical validity assessment of genes frequently tested on hereditary breast and ovarian cancer susceptibility sequencing panels. Genet. Med. 2018. [Google Scholar] [CrossRef]
- Colas, C.; Golmard, L.; de Pauw, A.; Caputo, S.M.; Stoppa-Lyonnet, D. “Decoding hereditary breast cancer” benefits and questions from multigene panel testing. Breast 2019, 45, 29–35. [Google Scholar] [CrossRef]
- del Valle, J.; Feliubadalo, L.; Nadal, M.; Teule, A.; Miro, R.; Cuesta, R.; Tornero, E.; Menendez, M.; Darder, E.; Brunet, J.; et al. Identification and comprehensive characterization of large genomic rearrangements in the BRCA1 and BRCA2 genes. Breast Cancer Res. Treat. 2010, 122, 733–743. [Google Scholar] [CrossRef]
- Smith, T.M.; Lee, M.K.; Szabo, C.I.; Jerome, N.; McEuen, M.; Taylor, M.; Hood, L.; King, M.C. Complete genomic sequence and analysis of 117 kb of human DNA containing the gene BRCA1. Genome Res. 1996, 6, 1029–1049. [Google Scholar] [CrossRef]
- Moisan, A.M.; Fortin, J.; Dumont, M.; Samson, C.; Bessette, P.; Chiquette, J.; Laframboise, R.; Lepine, J.; Lesperance, B.; Pichette, R.; et al. No evidence of BRCA1/2 genomic rearrangements in high-risk french-canadian breast/ovarian cancer families. Genet. Test. 2006, 10, 104–115. [Google Scholar] [CrossRef]
- Lahti-Domenici, J.; Rapakko, K.; Paakkonen, K.; Allinen, M.; Nevanlinna, H.; Kujala, M.; Huusko, P.; Winqvist, R. Exclusion of large deletions and other rearrangements in BRCA1 and BRCA2 in finnish breast and ovarian cancer families. Cancer Genet. Cytogenet. 2001, 129, 120–123. [Google Scholar] [CrossRef]
- Rouleau, E.; Jesson, B.; Briaux, A.; Nogues, C.; Chabaud, V.; Demange, L.; Sokolowska, J.; Coulet, F.; Barouk-Simonet, E.; Bignon, Y.J.; et al. Rare germline large rearrangements in the BRCA1/2 genes and eight candidate genes in 472 patients with breast cancer predisposition. Breast Cancer Res. Treat. 2012, 133, 1179–1190. [Google Scholar] [CrossRef]
- Montagna, M.; Dalla Palma, M.; Menin, C.; Agata, S.; De Nicolo, A.; Chieco-Bianchi, L.; D’Andrea, E. Genomic rearrangements account for more than one-third of the BRCA1 mutations in northern Italian breast/ovarian cancer families. Hum. Mol. Genet. 2003, 12, 1055–1061. [Google Scholar] [CrossRef]
- Peixoto, A.; Santos, C.; Rocha, P.; Pinheiro, M.; Principe, S.; Pereira, D.; Rodrigues, H.; Castro, F.; Abreu, J.; Gusmao, L.; et al. The c.156_157insalu BRCA2 rearrangement accounts for more than one-fourth of deleterious BRCA mutations in northern/central portugal. Breast Cancer Res. Treat. 2009, 114, 31–38. [Google Scholar] [CrossRef]
- Toland, A.E.; Forman, A.; Couch, F.J.; Culver, J.O.; Eccles, D.M.; Foulkes, W.D.; Hogervorst, F.B.L.; Houdayer, C.; Levy-Lahad, E.; Monteiro, A.N.; et al. Clinical testing of BRCA1 and BRCA2: A worldwide snapshot of technological practices. Npj Genom. Med. 2018, 3, 7. [Google Scholar] [CrossRef]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Fan, I.; Tang, J.; Li, S.; Zhang, S.; Shaw, P.A.; et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin-cohort study in Ontario, Canada. J. Natl. Cancer Inst. 2006, 98, 1694–1706. [Google Scholar] [CrossRef]
- Whittemore, A.S.; Gong, G.; John, E.M.; McGuire, V.; Li, F.P.; Ostrow, K.L.; Dicioccio, R.; Felberg, A.; West, D.W. Prevalence of BRCA1 mutation carriers among U.S. Non-hispanic whites. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2078–2083. [Google Scholar]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef]
- Struewing, J.P.; Hartge, P.; Wacholder, S.; Baker, S.M.; Berlin, M.; McAdams, M.; Timmerman, M.M.; Brody, L.C.; Tucker, M.A. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among ashkenazi jews. N. Engl. J. Med. 1997, 336, 1401–1408. [Google Scholar] [CrossRef]
- Peto, J.; Collins, N.; Barfoot, R.; Seal, S.; Warren, W.; Rahman, N.; Easton, D.F.; Evans, C.; Deacon, J.; Stratton, M.R. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J. Natl. Cancer Inst. 1999, 91, 943–949. [Google Scholar] [CrossRef]
- Kurian, A.W. brca1 and BRCA2 mutations across race and ethnicity: Distribution and clinical implications. Curr. Opin. Obstet. Gynecol. 2010, 22, 72–78. [Google Scholar] [CrossRef]
- Lesueur, F.; Mebirouk, N.; Jiao, Y.; Barjhoux, L.; Belotti, M.; Laurent, M.; Leone, M.; Houdayer, C.; Bressac-de Paillerets, B.; Vaur, D.; et al. Gemo, a national resource to study genetic modifiers of breast and ovarian cancer risk in BRCA1 and BRCA2 pathogenic variant carriers. Front. Oncol. 2018, 8, 490. [Google Scholar] [CrossRef]
- Caputo, S.; Benboudjema, L.; Sinilnikova, O.; Rouleau, E.; Beroud, C.; Lidereau, R. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the umd-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012, 40, D992–D1002. [Google Scholar] [CrossRef]
- Oros, K.K.; Ghadirian, P.; Maugard, C.M.; Perret, C.; Paredes, Y.; Mes-Masson, A.M.; Foulkes, W.D.; Provencher, D.; Tonin, P.N. Application of BRCA1 and BRCA2 mutation carrier prediction models in breast and/or ovarian cancer families of french canadian descent. Clin. Genet. 2006, 70, 320–329. [Google Scholar] [CrossRef]
- Tonin, P.; Weber, B.; Offit, K.; Couch, F.; Rebbeck, T.R.; Neuhausen, S.; Godwin, A.K.; Daly, M.; Wagner-Costalos, J.; Berman, D.; et al. Frequency of recurrent BRCA1 and BRCA2 mutations in ashkenazi jewish breast cancer families. Nat. Med. 1996, 2, 1179–1183. [Google Scholar] [CrossRef]
- Tonin, P.N.; Perret, C.; Lambert, J.A.; Paradis, A.J.; Kantemiroff, T.; Benoit, M.H.; Martin, G.; Foulkes, W.D.; Ghadirian, P. Founder brca1 and brca2 mutations in early-onset french canadian breast cancer cases unselected for family history. Int. J. Cancer 2001, 95, 189–193. [Google Scholar] [CrossRef]
- Gorski, B.; Byrski, T.; Huzarski, T.; Jakubowska, A.; Menkiszak, J.; Gronwald, J.; Pluzanska, A.; Bebenek, M.; Fischer-Maliszewska, L.; Grzybowska, E.; et al. Founder mutations in the brca1 gene in polish families with breast-ovarian cancer. Am. J. Hum. Genet. 2000, 66, 1963–1968. [Google Scholar] [CrossRef]
- Villarreal-Garza, C.; Weitzel, J.N.; Llacuachaqui, M.; Sifuentes, E.; Magallanes-Hoyos, M.C.; Gallardo, L.; Alvarez-Gomez, R.M.; Herzog, J.; Castillo, D.; Royer, R.; et al. The prevalence of BRCA1 and BRCA2 mutations among young mexican women with triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 150, 389–394. [Google Scholar] [CrossRef]
- Ossa, C.A.; Torres, D. Founder and recurrent mutations in brca1 and brca2 genes in latin american countries: State of the art and literature review. Oncologist 2016, 21, 832–839. [Google Scholar] [CrossRef]
- Manchanda, R.; Patel, S.; Antoniou, A.C.; Levy-Lahad, E.; Turnbull, C.; Evans, D.G.; Hopper, J.L.; Macinnis, R.J.; Menon, U.; Jacobs, I.; et al. Cost-effectiveness of population based BRCA testing with varying ashkenazi jewish ancestry. Am. J. Obs. Gynecol. 2017, 217, 578.e1–578.e12. [Google Scholar] [CrossRef]
- Lynce, F.; Isaacs, C. Population-based brca1/2 testing in ashkenazi jews: Ready for prime time. J. Natl. Compr. Cancer Netw. 2016, 14, 809–812. [Google Scholar] [CrossRef]
- King, M.C.; Levy-Lahad, E.; Lahad, A. Population-based screening for BRCA1 and BRCA2: 2014 lasker award. JAMA 2014, 312, 1091–1092. [Google Scholar] [CrossRef]
- Easton, D.F.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wenstrup, R.J.; len-Brady, K.; Tavtigian, S.V.; Monteiro, A.N.; Iversen, E.S.; Couch, F.J.; et al. A systematic genetic assessment of 1433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am. J. Hum. Genet. 2007, 81, 873–883. [Google Scholar] [CrossRef]
- Goldgar, D.E.; Easton, D.F.; Deffenbaugh, A.M.; Monteiro, A.N.; Tavtigian, S.V.; Couch, F.J.; Breast Cancer Information Core Steering Committee. Integrated evaluation of DNA sequence variants of unknown clinical significance: Application to BRCA1 and BRCA2. Am. J. Hum. Genet. 2004, 75, 535–544. [Google Scholar] [CrossRef]
- Goldgar, D.E.; Easton, D.F.; Byrnes, G.B.; Spurdle, A.B.; Iversen, E.S.; Greenblatt, M.S. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008, 29, 1265–1272. [Google Scholar] [CrossRef]
- Eggington, J.M.; Bowles, K.R.; Moyes, K.; Manley, S.; Esterling, L.; Sizemore, S.; Rosenthal, E.; Theisen, A.; Saam, J.; Arnell, C.; et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin. Genet. 2014, 86, 229–237. [Google Scholar] [CrossRef]
- Pesaran, T.; Karam, R.; Huether, R.; Li, S.; Farber-Katz, S.; Chamberlin, A.; Chong, H.; LaDuca, H.; Elliott, A. Beyond DNA: An integrated and functional approach for classifying germline variants in breast cancer genes. Int. J. Breast Cancer 2016, 2016, 2469523. [Google Scholar] [CrossRef]
- Lovelock, P.K.; Spurdle, A.B.; Mok, M.T.; Farrugia, D.J.; Lakhani, S.R.; Healey, S.; Arnold, S.; Buchanan, D.; Investigators, K.; Couch, F.J.; et al. Identification of brca1 missense substitutions that confer partial functional activity: Potential moderate risk variants? BCR 2007, 9, R82. [Google Scholar] [CrossRef]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallee, M.P.; Monteiro, A.N.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (vus). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef]
- Eccles, D.M.; Mitchell, G.; Monteiro, A.N.; Schmutzler, R.; Couch, F.J.; Spurdle, A.B.; Gomez-Garcia, E.B.; Group, E.C.W. BRCA1 and BRCA2 genetic testing-pitfalls and recommendations for managing variants of uncertain clinical significance. Ann. Oncol. 2015, 26, 2057–2065. [Google Scholar] [CrossRef]
- Szabo, C.I.; Worley, T.; Monteiro, A.N. Understanding germ-line mutations in BRCA1. Cancer Biol. Ther. 2004, 3, 515–520. [Google Scholar] [CrossRef][Green Version]
- de la Hoya, M.; Soukarieh, O.; Lopez-Perolio, I.; Vega, A.; Walker, L.C.; van Ierland, Y.; Baralle, D.; Santamarina, M.; Lattimore, V.; Wijnen, J.; et al. Combined genetic and splicing analysis of BRCA1 c.[594-2a>c; 641a>g] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum. Mol. Genet. 2016, 25, 2256–2268. [Google Scholar] [CrossRef]
- Friedman, L.S.; Ostermeyer, E.A.; Szabo, C.I.; Dowd, P.; Lynch, E.D.; Rowell, S.E.; King, M.C. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat. Genet. 1994, 8, 399–404. [Google Scholar] [CrossRef]
- Gough, C.A.; Gojobori, T.; Imanishi, T. Cancer-related mutations in BRCA1-BRCT cause long-range structural changes in protein-protein binding sites: A molecular dynamics study. Proteins 2007, 66, 69–86. [Google Scholar] [CrossRef]
- Monteiro, A.N.; August, A.; Hanafusa, H. Evidence for a transcriptional activation function of brca1 c-terminal region. Proc. Natl. Acad. Sci. USA 1996, 93, 13595–13599. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Au, W.W.; Henderson, B.R. Cytoplasmic mislocalization of BRCA1 caused by cancer-associated mutations in the BRCT domain. Exp. Cell Res. 2004, 293, 14–21. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Jiang, Q.; Greenberg, R.A. Deciphering the BRCA1 tumor suppressor network. J. Biol. Chem. 2015, 290, 17724–17732. [Google Scholar] [CrossRef]
- Jhuraney, A.; Velkova, A.; Johnson, R.C.; Kessing, B.; Carvalho, R.S.; Whiley, P.; Spurdle, A.B.; Vreeswijk, M.P.; Caputo, S.M.; Millot, G.A.; et al. Brca1 circos: A visualisation resource for functional analysis of missense variants. J. Med. Genet. 2015, 52, 224–230. [Google Scholar] [CrossRef]
- Millot, G.A.; Carvalho, M.A.; Caputo, S.M.; Vreeswijk, M.P.; Brown, M.A.; Webb, M.; Rouleau, E.; Neuhausen, S.L.; Hansen, T.; Galli, A.; et al. A guide for functional analysis of BRCA1 variants of uncertain significance. Hum. Mutat. 2012, 33, 1526–1537. [Google Scholar] [CrossRef]
- Fernandes, V.C.G.; Golubeva, V.A.; Di Pietro, G.; Shields, C.; Amankwah, K.; Nepomuceno, T.C.; de Gregoriis, G.; Abreu, R.B.; Harro, C.; Gomes, T.T.; et al. Impact of amino acid substitutions at secondary structures in the BRCT domains of the tumor suppressor BRCA1: Implications for clinical annotation. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Woods, N.T.B.R.; Golubeva, V.; Jhuraney, A.; De-Gregoriis, G.; Vaclova, T.; Goldgar, D.E.; Couch, F.J.; Carvalho, M.A.; Iversen, E.S.; Monteiro, A.N. Functional assays provide a robust tool for the clinical annotation of genetic variants of uncertain significance. NPJ Genom. Med. 2016, 1, 16001. [Google Scholar] [CrossRef]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Starita, L.M.; Islam, M.M.; Banerjee, T.; Adamovich, A.I.; Gullingsrud, J.; Fields, S.; Shendure, J.; Parvin, J.D. A multiplex homology-directed DNA repair assay reveals the impact of more than 1000 BRCA1 missense substitution variants on protein function. Am. J. Hum. Genet. 2018, 103, 498–508. [Google Scholar] [CrossRef]
- Szabo, C.; Masiello, A.; Ryan, J.F.; Brody, L.C. The breast cancer information core: Database design, structure, and scope. Hum. Mutat. 2000, 16, 123–131. [Google Scholar] [CrossRef]
- Friend, S.; Borresen, A.L.; Brody, L.; Casey, G.; Devilee, P.; Gayther, S.; Goldgar, D.; Murphy, P.; Weber, B.L.; Wiseman, R. Breast cancer information on the web. Nat. Genet. 1995, 11, 238–239. [Google Scholar] [CrossRef]
- Toland, A.E.; Brody, L.C.; BIC Steering Committee. Lessons learned from two decades of BRCA1 and BRCA2 genetic testing: The evolution of data sharing and variant classification. Genet. Med. 2018. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.; Monteiro, A.N.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA—Evidence-based network for the interpretation of germline mutant alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef]
- Tan, E.C.; Loh, M.; Chuon, D.; Lim, Y.P. Singapore human mutation/polymorphism database: A country-specific database for mutations and polymorphisms in inherited disorders and candidate gene association studies. Hum. Mutat. 2006, 27, 232–235. [Google Scholar] [CrossRef]
- Osborne, R.H.; Hopper, J.L.; Kirk, J.A.; Chenevix-Trench, G.; Thorne, H.J.; Sambrook, J.F. Kconfab: A research resource of australasian breast cancer families. Kathleen cuningham foundation consortium for research into familial breast cancer. Med. J. Aust. 2000, 172, 463–464. [Google Scholar]
- Beroud, C.; Letovsky, S.I.; Braastad, C.D.; Caputo, S.M.; Beaudoux, O.; Bignon, Y.J.; Bressac-De Paillerets, B.; Bronner, M.; Buell, C.M.; Collod-Beroud, G.; et al. BRCA share: A collection of clinical Brca gene variants. Hum. Mutat. 2016, 37, 1318–1328. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of brazilians in the abraom database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golubeva, V.A.; Nepomuceno, T.C.; Monteiro, A.N.A. Germline Missense Variants in BRCA1: New Trends and Challenges for Clinical Annotation. Cancers 2019, 11, 522. https://doi.org/10.3390/cancers11040522
Golubeva VA, Nepomuceno TC, Monteiro ANA. Germline Missense Variants in BRCA1: New Trends and Challenges for Clinical Annotation. Cancers. 2019; 11(4):522. https://doi.org/10.3390/cancers11040522
Chicago/Turabian StyleGolubeva, Volha A., Thales C. Nepomuceno, and Alvaro N. A. Monteiro. 2019. "Germline Missense Variants in BRCA1: New Trends and Challenges for Clinical Annotation" Cancers 11, no. 4: 522. https://doi.org/10.3390/cancers11040522
APA StyleGolubeva, V. A., Nepomuceno, T. C., & Monteiro, A. N. A. (2019). Germline Missense Variants in BRCA1: New Trends and Challenges for Clinical Annotation. Cancers, 11(4), 522. https://doi.org/10.3390/cancers11040522