Membrane Lipidome Reorganization and Accumulation of Tissue DNA Lesions in Tumor-Bearing Mice: An Exploratory Study




 ,
,  and
and 
Abstract
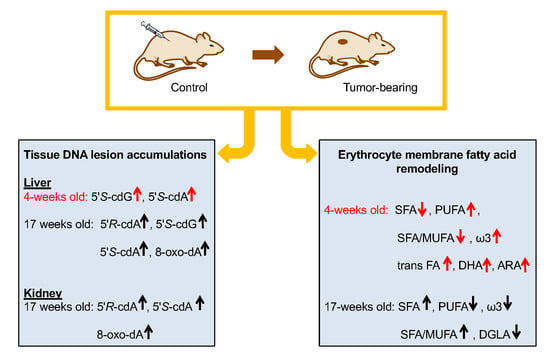
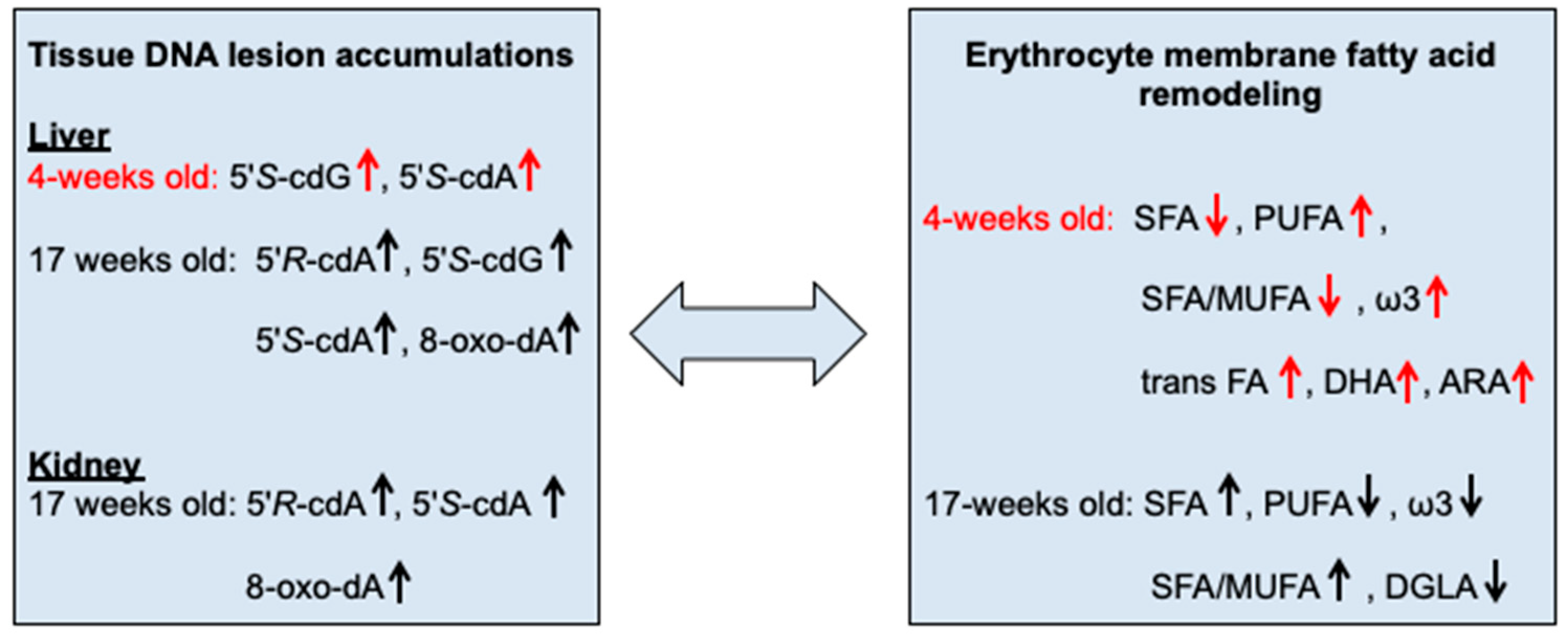
:
1. Introduction
2. Results
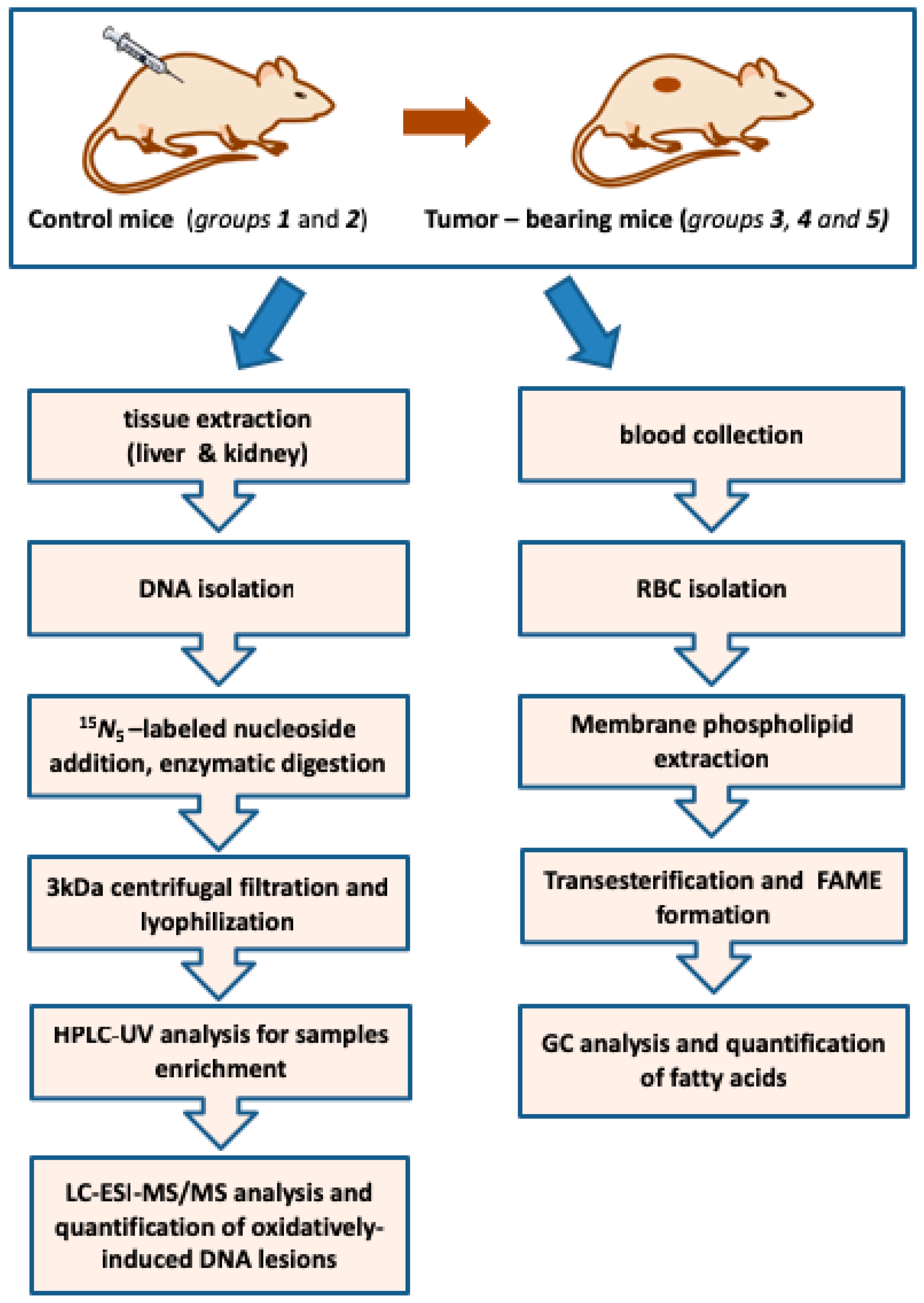
2.1. Protocol Outline
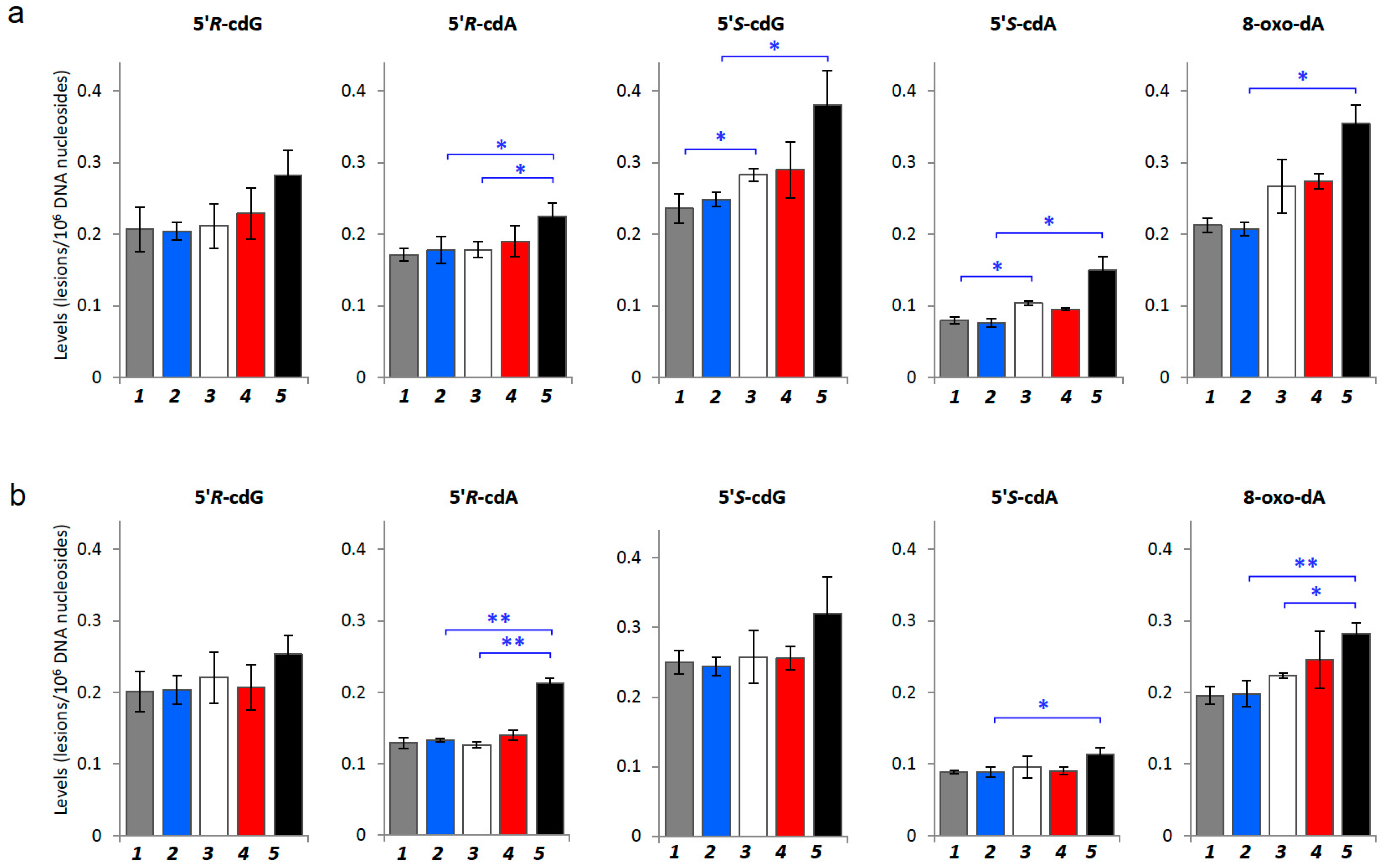
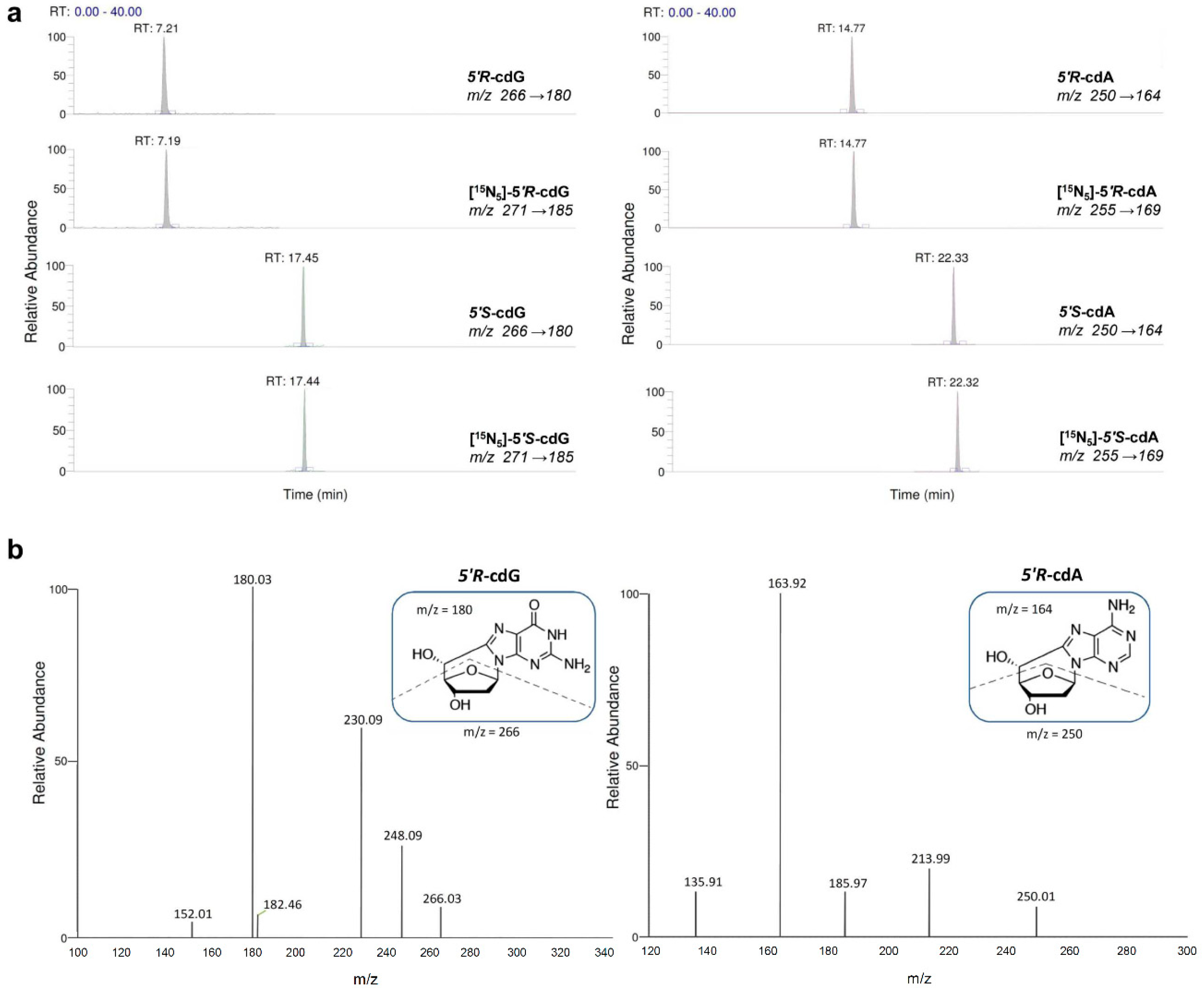
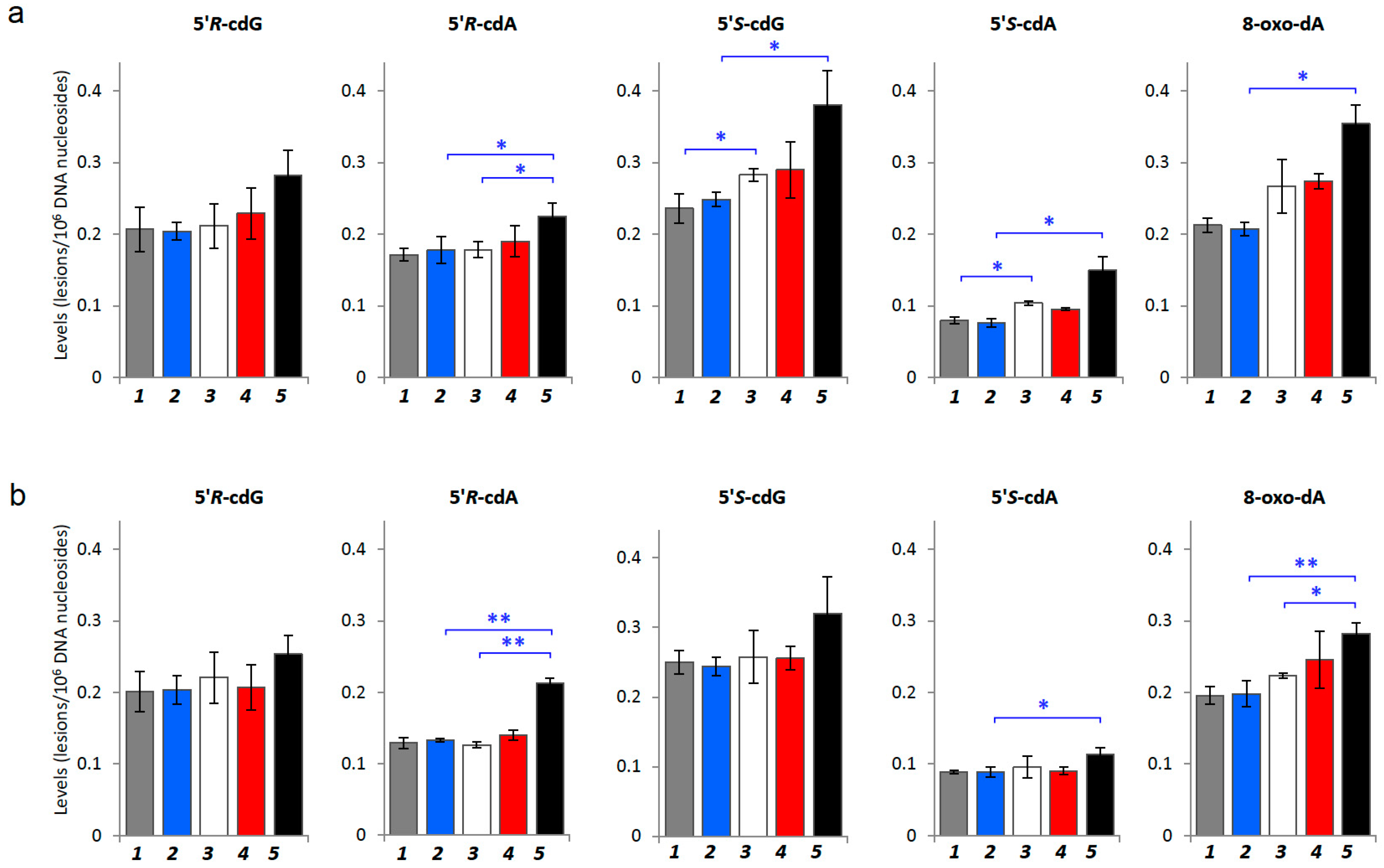
2.2. Oxidatively-Induced DNA Lesions Accumulation in Organs
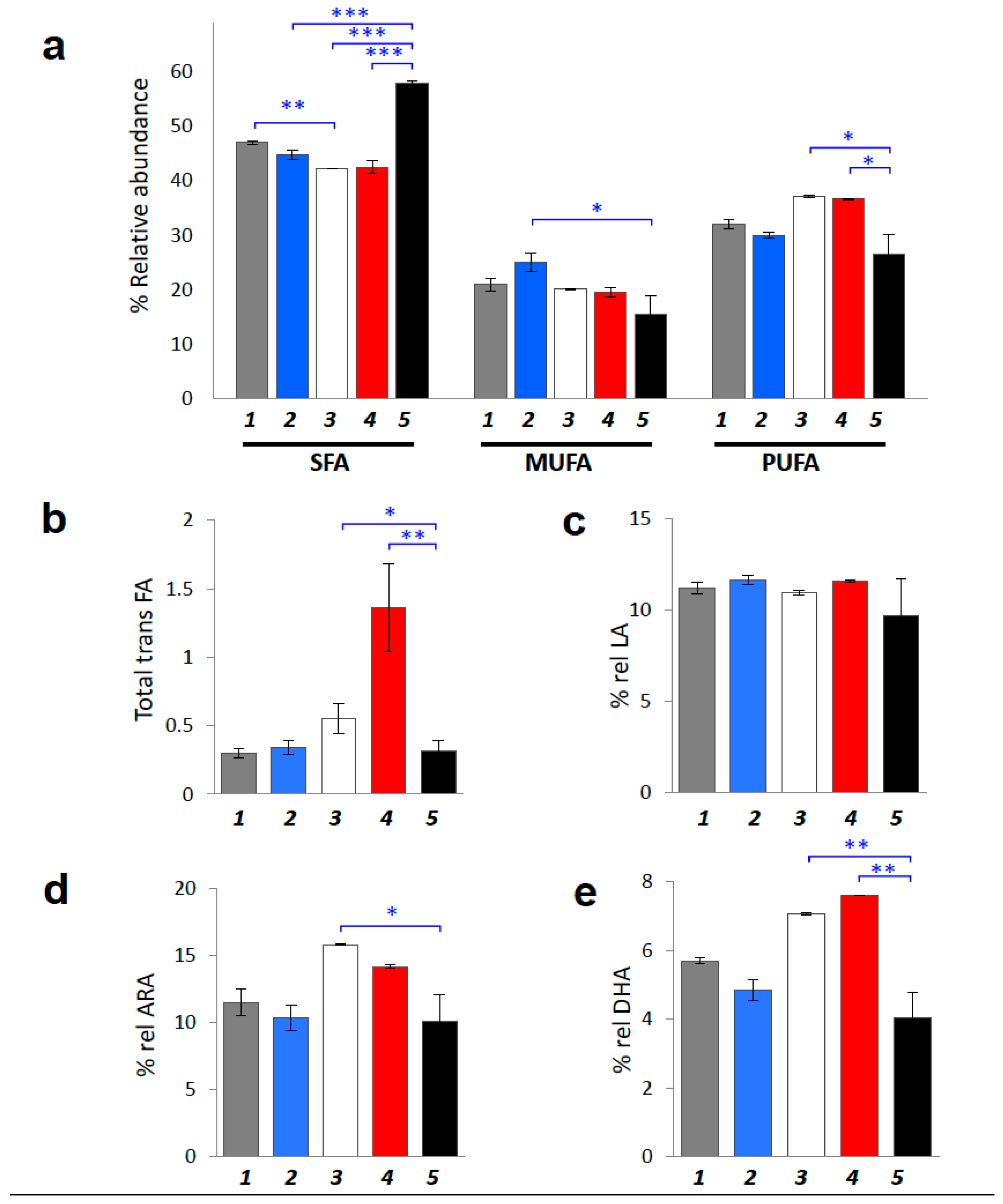
2.3. Fatty Acid-Based Membrane Lipidomics of Erythrocytes
- (A)
- Membrane lipidome profiles showed PUFA level decrease during tumor propagation. 17-weeks-old tumor-bearing mice are characterized by significant less PUFA content compared to the young (4- and 5-weeks-old) tumor-bearing age mice (p = 0.0071 and p = 0.0085, respectively). Tumor-bearing mice at early ages showed increased PUFA levels, becoming similar to healthy Swiss mice (compare Table 4 with Table S4). At 17-weeks of age tumor-bearing SCID mice showed the significant depletion of PUFA in RBC membrane phospholipids (p = 0.0019) with a corresponding increase of SFA (p < 0.0001). In particular, the relative percentage of SFA reached ca 58% over the total fatty acid composition in late stage tumor-bearing mice, the highest SFA percentage found compared to both control SCID and healthy Swiss mice (Table 4 and Table S4), and also compared to the percentage of SFA that corresponds to early stage tumor-bearing mice (ca. 42%; (p < 0.0001)).
- (B)
- Some members of the ω-6 and ω-3 PUFA families (ARA and DHA graphically represented in Figure 4d,e, respectively) are also interesting membrane components to follow, showing significant depletion in tumor-bearing SCID mice after 17 weeks progress (p = 0.0333 and p = 0.0027, respectively). Remarkably, in all cases the PUFA changes did not involve the ω-6 precursor linoleic acid (LA: 9cis,12cis-C18:2), showing only a diminution trend at the late tumor stage (Table 3).
- (C)
- Indexes of unsaturation and peroxidation are significantly decreased (UI and PI; p = 0.0014 and p = 0.0053, respectively). It is worth noting that neither in SCID mice nor in healthy Swiss mice the UI and PI indices were found significantly affected by the age (see Table 4 and Table S4). Moreover, in SCID mice of 4-weeks these indices were more comparable to the Swiss healthy than to the SCID mice (cfr., Table 4 with Table S4).
- (D)
- The level of TFA in tumor-bearing SCID mice, was significantly increased at 5-weeks and again decreased in 17-weeks old tumor-bearing mice (p = 0.0074) (Table 4 and Figure 4c). No significant changes were revealed for control SCID mice while only in healthy Swiss mice the TFA were decreased during ageing (Table S4, p = 0.0006).
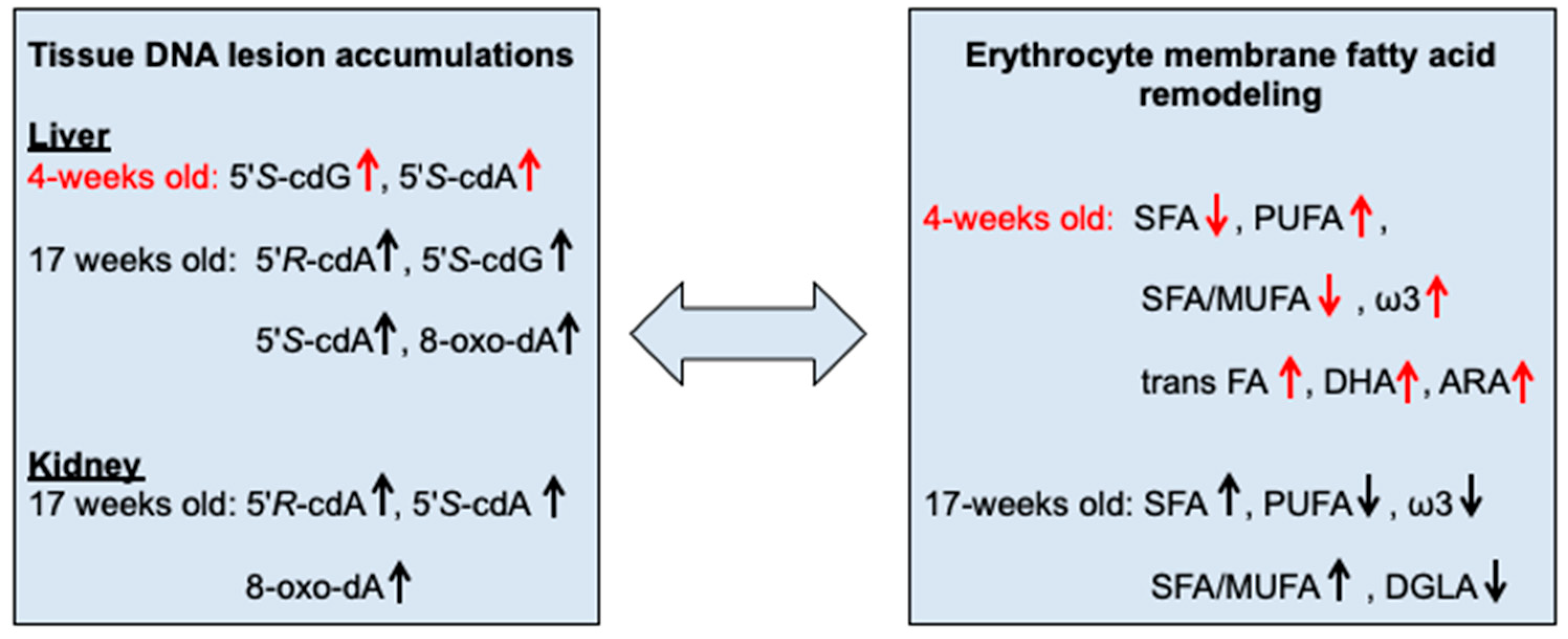
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Studies
4.3. Cell Lines, Cell Culture Conditions and Xenograft Construction
4.4. Genomic DNA Isolation
4.5. Enzymatic Digestion to Nucleosides
4.6. Analysis by High Performance Liquid Chromatography, Clean-Up and Enrichment
4.7. Measurement of Modified Nucleosides by LC-ESI-MS/MS
4.8. RBC Fatty Acid-Based Membrane Lipidomic Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 16:0 | palmitic acid |
| 18:0 | stearic acid |
| ARA | arachidonic acid |
| BER | base excision repair |
| cdA | 5′ 8-cyclo-2′-deoxyadenosine |
| cdG | 5′ 8-cyclo-2′-deoxyguanosine |
| cPu | purine 5′ 8-cyclo-2′-deoxynucleoside |
| DHA | docosahexaenoic acid |
| DGLA | dihomo-gamma-linolenic acid |
| EPA | eicosapentaenoic acid |
| FAME | fatty acid methyl esters |
| FASN | fatty acid synthase |
| GC | gas chromatography |
| LA | linoleic acid |
| LC-MS/MS | liquid chromatography tandem mass spectrometry |
| MUFA | monounsaturated fatty acids |
| NER | nucleotide excision repair |
| 8-oxo-dA | 8-oxo-7,8-dihydro-2′-deoxyadenosine |
| PI | peroxidation index |
| PUFA | polyunsaturated fatty acids |
| RBC | red blood cells |
| ROS | reactive oxygen species |
| SCID | severe combined immunodeficient |
| SFA | saturated fatty acids |
| TFA | trans fatty acids |
| UI | unsaturation index |
References
- Campisi, J. Aging, cellular senescence, and cancer. Ann. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Gladyshev, V. The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancercells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, K.; Jose, L.; McFaline, L.J.; Pang, B.; Sullivan, M.; Dong, M.; Plummer, E.; Dedon, P.C. Quantification of DNA damage products resulting from deamination, oxidation anreaction with products of lipid peroxidation by liquid chromatography isotope dilutiontandem mass spectrometry. Nat. Protoc. 2008, 3, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cui, Y.; Niedernhofer, L.J.; Wang, Y. Occurrence, biological consequences, and human health relevance of oxidative stress-induced DNA damage. Chem. Res. Toxicol. 2016, 29, 2008–2039. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Terzidis, M.A. Purine 5′,8-cyclonucleoside lesions: Chemistry and biology. Chem. Soc. Rev. 2011, 40, 1368–1382. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Krokidis, M.G.; Papadopoulos, K.; Terzidis, M.A. Purine 5′,8-cyclo-2′-deoxynucleoside lesions in irradiated DNA. Radiat. Phys. Chem. 2016, 128, 75–81. [Google Scholar] [CrossRef]
- Brooks, P.J. The cyclopurine deoxynucleosides: DNA repair, biological effects, mechanistic insights, and unanswered questions. Free Radic. Biol. Med. 2017, 107, 90–100. [Google Scholar] [CrossRef]
- Brooks, P.J.; Wise, D.S.; Berry, D.A.; Kosmoski, J.V.; Smerdon, M.J.; Somers, R.L.; Mackie, H.; Spoonde, A.Y.; Ackerman, E.J.; Coleman, K.; et al. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 2000, 275, 22355–22362. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; Lindahl, T. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar] [CrossRef]
- Kropachev, K.; Ding, S.; Terzidis, M.A.; Masi, A.; Liu, Z.; Cai, Y.; Kolbanovskiy, M.; Chatgilialoglu, C.; Broyde, S.; Geacintov, N.E.; et al. Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 2014, 42, 5020–5032. [Google Scholar] [CrossRef]
- You, C.; Swanson, A.L.; Dai, X.; Yuan, B.; Wang, J.; Wang, Y. Translesion synthesis of 8,5′-cyclopurine-2′-deoxynucleosides by DNA polymerases η, ι and ζ. J. Biol. Chem. 2013, 288, 28548–28556. [Google Scholar] [CrossRef]
- Xu, W.; Ouellette, A.M.; Wawrzak, Z.; Shriver, S.J.; Anderson, S.M.; Zhao, L. Kinetic and structural mechanisms of (5′S)-8,5′-cyclo-2′-deoxyguanosine-induced DNA replication stalling. Biochemistry 2015, 54, 639–651. [Google Scholar] [CrossRef]
- Xu, M.; Lai, Y.; Jiang, Z.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. A 5′,8-cyclo-2′-deoxypurine lesion induces trinucleotide repeat deletion via a unique lesion bypass by DNA polymerase β. Nucleic Acids Res. 2014, 42, 13749–13763. [Google Scholar] [CrossRef]
- Jiang, Z.; Xu, M.; Lai, Y.; Laverde, E.E.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. Bypass of a 5′,8-cyclopurine-2′-deoxynucleoside by DNA polymerase β during DNA replication and base excision repair leads to nucleotide misinsertions and DNA strand breaks. DNA Repair 2015, 33, 24–34. [Google Scholar] [CrossRef]
- Anderson, K.M.; Jaruga, P.; Ramsey, C.R.; Gilman, N.K.; Green, V.M.; Rostad, S.W.; Emerman, J.T.; Dizdaroglu, M.; Malins, D.C. Structural alterations in breast stromal and epithelial DNA: The influence of 8,5′-cyclo-2′-deoxyadenosine. Cell Cycle 2006, 5, 1240–1244. [Google Scholar] [CrossRef]
- Nyaga, S.G.; Jaruga, P.; Lohani, A.; Dizdaroglu, M.; Evans, M.K. Accumulation of oxidatively induced DNA damage in human breast cancer cell lines following treatment with hydrogen peroxide. Cell Cycle 2007, 6, 1472–1478. [Google Scholar] [CrossRef]
- D’Errico, M.; Parlanti, E.; Teson, M.; Degan, P.; Lemma, T.; Calcagnile, A.; Iavarone, I.; Jaruga, P.; Ropolo, M.; Pedrini, A.M.; et al. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene 2007, 26, 4336–4343. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Coskun, E.; Jaruga, P. Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic. Res. 2015, 49, 525–548. [Google Scholar] [CrossRef]
- Terzidis, M.A.; Chatgilialoglu, C. An ameliorative protocol for the quantification of purine 5′,8-cyclo-2′-deoxynucleosides in oxidized DNA. Front. Chem. 2015, 3, 47. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Terzidis, M.A.; Efthimiadou, E.; Zervou, S.K.; Kordas, G.; Papadopoulos, K.; Hiskia, A.; Kletsas, D.; Chatgilialoglu, C. Purine 5′,8-cyclo-2′-deoxynucleoside lesions: Formation by radical stress and repair in human breast epithelial cancer cells. Free Radic. Res. 2017, 51, 470–482. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, P.; Cui, Y.; Wang, Y. Chemical analysis of DNA damage. Anal. Chem. 2018, 90, 556–576. [Google Scholar] [CrossRef]
- Wang, J.; Clauson, C.L.; Robbins, P.D.; Niedernhofer, L.J.; Wang, Y. The oxidative DNA lesions 8,5′ cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell 2012, 11, 714–716. [Google Scholar] [CrossRef]
- Wang, J.; Yuan, B.; Guerrero, C.; Bahde, R.; Gupta, S.; Wang, Y. Quantification of oxidative DNA lesions in tissues of Long-Evans Cinnamon rats by capillary high-performance liquid chromatography-tandem mass spectrometry coupled with stable isotope dilution method. Anal. Chem. 2011, 83, 2201–2209. [Google Scholar] [CrossRef]
- Yu, Y.; Guerrero, C.R.; Liu, S.; Amato, N.J.; Sharma, Y.; Gupta, S.; Wang, Y. Comprehensive assessment of oxidatively induced modifications of DNA in a rat model of human Wilson’s disease. Mol. Cell. Proteom. 2016, 15, 810–817. [Google Scholar] [CrossRef]
- Egler, R.A.; Fernandes, E.; Rothermund, K.; Sereika, S.; de Souza-Pinto, N.; Jaruga, P.; Dizdaroglu, M.; Prochownik, E.V. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene 2005, 24, 8038–8050. [Google Scholar] [CrossRef]
- Ferreri, C.; Masi, A.; Sansone, A.; Giacometti, G.; Larocca, A.V.; Menounou, G.; Scanferlato, R.; Tortorella, S.; Rota, D.; Conti, M.; et al. Fatty acids in membranes as homeostatic, metabolic and nutritional biomarkers: Recent advancements in analytics and diagnostics. Diagnostics 2017, 7, 1. [Google Scholar] [CrossRef]
- Ferreri, C.; Chatgilialoglu, C. Role of fatty acid-based functional lipidomics in the development of molecular diagnostic tools. Expert Rev. Mol. Diagn. 2012, 7, 767–780. [Google Scholar] [CrossRef]
- Coviello, G.; Tutino, V.; Notarnicola, M.; Caruso, M.G. Erythrocyte membrane fatty acids profile in colorectal cancer patients: A preliminary study. Anticancer Res. 2014, 34, 4775–4779. [Google Scholar]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Igal, R.A. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1865–1880. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Comba, A.; Lin, Y.H.; Eynard, A.R.; Valentich, M.A.; Fernandez-Zapico, M.E.; Pasqualini, M.E. Basic aspects of tumor cell fatty acid-regulated signaling and transcription factors. Cancer Metastasis Rev. 2011, 30, 325–342. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Tosi, F.; Sartori, F.; Guarini, P.; Olivieri, O.; Martinelli, N. Delta-5 and delta-6 desaturases: Crucial enzymes in polyunsaturated fatty acid-related pathways with pleiotropic influences in health and disease. Adv. Exp. Med. Biol. 2014, 824, 61–81. [Google Scholar]
- Robichaud, P.P.; Surette, M.E. Polyunsaturated fatty acid–phospholipid remodeling and inflammation. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 112–118. [Google Scholar] [CrossRef]
- Marini, M.; Abruzzo, P.M.; Bolotta, A.; Veicsteinas, A.; Ferreri, C. Aerobic training affects fatty acid composition of erythrocyte membranes. Lipids Health Dis. 2011, 10, 188. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of glycerolipides; a comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Melchiorre, M.; Sansone, A.; Torreggiani, A. Lipid geometrical isomerism: From chemistry to biology and diagnostics. Chem. Rev. 2014, 114, 255–284. [Google Scholar] [CrossRef]
- Dranoff, G. Experimental mouse tumor models: What can be learnt about human cancer immunology. Nat. Rev. Immunol. 2011, 12, 61–66. [Google Scholar] [CrossRef]
- Morton, C.L.; Houghton, P.J. Establishment of human tumor xenografts in immunodeficient mice. Nat. Protoc. 2007, 2, 247–250. [Google Scholar] [CrossRef]
- Robinson, A.R.; Yousefzadeh, M.J.; Rozgaja, T.A.; Wang, J.; Li, X.; Tilstra, J.S.; Feldman, C.H.; Gregg, S.Q.; Johnson, C.H.; Skoda, E.M.; et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 2018, 17, 259–273. [Google Scholar] [CrossRef]
- Traube, F.R.; Schiffers, S.; Iwan, K.; Kellner, S.; Spada, F.; Müller, M.; Carell, T. Isotope-dilution mass spectrometry for exact quantification of noncanonical DNA nucleosides. Nat. Protoc. 2019, 14, 283–312. [Google Scholar] [CrossRef]
- Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Kareva, I.G.; Naf, D.; Nowsheen, S.; Kryston, T.B.; Bonner, W.M.; Georgakilas, A.G.; Sedelnikova, O.A. Tumors induce complex DNA damage in distant proliferative tissues in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17992–17997. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef]
- Thompson, H.J. Oxidative DNA damage and cancer risk assessment. J. Nutr. 2006, 136, S2693–S2694. [Google Scholar] [CrossRef]
- Rodriguez, H.; Jaruga, P.; Leber, D.; Nyaga, S.G.; Evans, M.K.; Dizdaroglu, M. Lymphoblasts of women with BRCA1 mutations are deficient in cellular repair of 8,5′-Cyclopurine-2′-deoxynucleosides and 8-hydroxy-2′-deoxyguanosine. Biochemistry 2007, 46, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Viviani Anselmi, C.; Ferreri, C.; Novelli, V.; Roncarati, R.; Bronzini, R.; Marchese, G.; Somalvico, F.; Condorelli, G.; Sandro Montenero, A.; Puca, A.A. Fatty acid percentage in erythrocyte membranes of atrial flutter/fibrillation patients and controls. J. Interv. Card. Electrophysiol. 2010, 27, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Puca, A.A.; Andrew, P.; Novelli, V.; Anselmi, C.V.; Somalvico, F.; Cirillo, N.A.; Chatgilialoglu, C.; Ferreri, C. Lipid profile of erythrocyte membranes as possible biomarker of longevity. Rejuven. Res. 2008, 11, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Prasinou, P.; Dafnis, I.; Giacometti, G.; Ferreri, C.; Chroni, A.; Chatgilialoglu, C. Fatty acid-based lipidomics and membrane remodelling induced by apoE3 and apoE4 in human neuroblastoma cells. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1967–1973. [Google Scholar] [CrossRef]
- De Magahaes, J.P. How ageing processes influence cancer. Nat. Rev. Cancer 2013, 13, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Chatgilialoglu, C. Membrane Lipidomics for Personalized Health; John Wiley & Sons: Chichester, UK, 2015. [Google Scholar]
- Yehuda, S.; Rabinovitz, S.; Mostofsky, D.I. Mediation of cognitive function by high diet following stress and inflammation. Nutr. Neurosci. 2005, 8, 309–315. [Google Scholar] [CrossRef]
- Azrad, M.; Turgeon, C.; Demark-Wahnefrid, W. Current evidence linking polyunsaturated fatty acids with cancer risk and progression. Front. Oncol. 2013, 3, 224. [Google Scholar] [CrossRef]
- Zheng, H.; Tang, H.; Liu, M.; He, M.; Lai, P.; Dong, H.; Lin, J.; Jia, C.; Zhong, M.; Dai, Y.; et al. Inhibition of endometrial cancer by n-3 polyunsaturated fatty acids in preclinical models. Cancer Prev. Res. 2014, 7, 824–834. [Google Scholar] [CrossRef]
- Lauritzen, L.; Hansen, H.S.; Jorgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Progr. Lipids Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Fujii, K.; Luo, Y.; Fujiwara-Tani, R.; Kishi, S.; He, S.; Yang, S.; Sasaki, T.; Ohmori, H.; Kuniyasu, H. Pro-metastatic intracellular signaling of the elaidic trans fatty acid. Int. J. Oncol. 2016, 50, 85–92. [Google Scholar] [CrossRef]
- Bell, E.; Ponthan, F.; Whitworth, C.; Westermann, F.; Thomas, H.; Redfern, C.P. Cell Survival signalling through PPARδ and arachidonic acid metabolites in neuroblastoma. PLoS ONE 2013, 8, e68859. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef]
- Tsiapa, I.; Efthimiadou, E.K.; Fragogeorgi, E.; Loudos, G.; Varvarigou, A.D.; Bouziotis, P.; Kordas, G.C.; Mihailidis, D.; Nikiforidis, G.C.; Xanthopoulos, S.; et al. 99mTc-labeled aminosilane-coated iron oxide nanoparticles for molecular imaging of ανβ3-mediated tumor expression and feasibility for hyperthermia treatment. J. Colloid Interface Sci. 2014, 433, 163–175. [Google Scholar] [CrossRef]
- Efthimiadou, E.K.; Fragogeorgi, E.; Palamaris, L.; Karampelas, T.; Lelovas, P.; Loudos, G.; Tamvakopoulos, C.; Kostomitsopoulos, N.; Kordas, G. Versatile quarto stimuli nanostructure based on Trojan Horse approach for cancer therapy: Synthesis, characterization, in vitro and in vivo studies. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 79, 605–612. [Google Scholar] [CrossRef]
- Terzidis, M.A.; Ferreri, C.; Chatgilialoglu, C. Radiation induced formation of purine lesions in single and double stranded DNA: Revised quantification. Front. Chem. 2015, 3, 18. [Google Scholar] [CrossRef]
- Terzidis, M.A.; Prisecaru, A.; Molphy, Z.; Barron, N.; Randazzo, A.; Dumont, E.; Krokidis, M.G.; Kellett, A.; Chatgilialoglu, C. Radical-induced purine lesion formation is dependent on DNA helical topology. Free Radic. Res. 2016, 50, S91–S101. [Google Scholar] [CrossRef]
- Sansone, A.; Melchiorre, M.; Chatgilialoglu, C.; Ferreri, C. Hexadecenoic fatty acid isomers: A chemical biology approach for human plasma biomarker development. Chem. Res. Toxicol. 2013, 26, 1703–1709. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice (Age, Week) | 5′R-cdG | 5′R-cdA | 5′S-cdG | 5′S-cdA | 8-Oxo-dA |
|---|---|---|---|---|---|
| Liver | |||||
| Control (4 w) | 0.207 ± 0.031 | 0.171 ± 0.009 | 0.236 ± 0.021 | 0.079 ± 0.005 | 0.212 ± 0.010 |
| Control (17 w) | 0.204 ± 0.012 | 0.178 ± 0.019 | 0.248 ± 0.010 | 0.076 ± 0.006 | 0.207 ± 0.009 |
| Tumor-bearing (4 w) | 0.211 ± 0.031 | 0.178 ± 0.011 | 0.283 ± 0.009 | 0.103 ± 0.003 | 0.267 ± 0.037 |
| Tumor-bearing (5 w) | 0.229 ± 0.035 | 0.190 ± 0.021 | 0.290 ± 0.039 | 0.095 ± 0.002 | 0.274 ± 0.011 |
| Tumor-bearing (17 w) | 0.282 ± 0.035 | 0.225 ± 0.018 | 0.381 ± 0.047 | 0.149 ± 0.020 | 0.354 ± 0.027 |
| Kidney | |||||
| Control (4 w) | 0.201 ± 0.028 | 0.129 ± 0.008 | 0.250 ± 0.017 | 0.089 ± 0.002 | 0.196 ± 0.012 |
| Control (17 w) | 0.204 ± 0.020 | 0.133 ± 0.003 | 0.244 ± 0.013 | 0.089 ± 0.007 | 0.198 ± 0.018 |
| Tumor-bearing (4 w) | 0.221 ± 0.036 | 0.126 ± 0.004 | 0.258 ± 0.038 | 0.095 ± 0.015 | 0.223 ± 0.003 |
| Tumor-bearing (5 w) | 0.207 ± 0.031 | 0.140 ± 0.007 | 0.256 ± 0.017 | 0.090 ± 0.006 | 0.246 ± 0.040 |
| Tumor-bearing (17 w) | 0.254 ± 0.027 | 0.213 ± 0.006 | 0.320 ± 0.053 | 0.113 ± 0.010 | 0.283 ± 0.015 |
| Mice (Age, Week) | 5′R-cdG | 5′R-cdA | 5′S-cdG | 5′S-cdA | 8-Oxo-dA |
|---|---|---|---|---|---|
| Liver | |||||
| Control (4 w) vs. Tumor-bearing (4 w) | 0.880 | 0.517 | 0.040 | 0.006 | 0.293 |
| Control (17 w) vs. Tumor-bearing (17 w) | 0.067 | 0.036 | 0.041 | 0.025 | 0.012 |
| Tumor-bearing (4 w) vs. Tumor-bearing (5 w) | 0.599 | 0.482 | 0.793 | 0.060 | 0.841 |
| Tumor-bearing (4 w) vs. Tumor-bearing (17 w) | 0.141 | 0.037 | 0.072 | 0.057 | 0.104 |
| p value (ANOVA) | 0.0568 | 0.0237 | 0.0022 | <0.0001 | <0.0001 |
| Kidney | |||||
| Control (4 w) vs. Tumor-bearing (4 w) | 0.571 | 0.663 | 0.836 | 0.651 | 0.065 |
| Control (17 w) vs. Tumor-bearing (17 w) | 0.059 | 0.000 | 0.136 | 0.025 | 0.003 |
| Tumor-bearing (4 w) vs. Tumor-bearing (5 w) | 0.699 | 0.073 | 0.955 | 0.734 | 0.438 |
| Tumor-bearing (4 w) vs. Tumor-bearing (17 w) | 0.383 | 0.000 | 0.222 | 0.289 | 0.022 |
| p value (ANOVA) | 0.2107 | <0.0001 | 0.0768 | 0.0244 | 0.0055 |
| FAME | Control (4 w) | Control (17 w) | Tumor-Bearing (4 w) | Tumor-Bearing (5 w) | Tumor-Bearing (17 w) | p Value |
|---|---|---|---|---|---|---|
| 14:0 | 0.57 ± 0.06 | 0.55 ± 0.14 | 0.31 ± 0.02 | 0.33 ± 0.02 | 0.54 ± 0.05 | 0.0103 |
| 15:0 | 0.15 ± 0.04 | 0.15 ± 0.02 | 0.36 ± 0.05 | 0.46 ± 0.06 | 0.09 ± 0.07 | 0.0142 |
| 16:0 | 35.38 ± 0.46 | 31.78 ± 0.55 | 28.61 ± 0.07 | 29.23 ± 0.91 | 38.52 ± 1.90 | 0.0053 |
| 16:1c6 | 0.23 ± 0.02 | 0.23 ± 0.05 | 0.26 ± 0.01 | 0.37 ± 0.08 | 0.22 ± 0.07 | 0.2394 |
| 16:1c9 | 1.66 ± 0.36 | 2.89 ± 0.38 | 1.47 ± 0.21 | 1.37 ± 0.05 | 1.15 ± 0.65 | 0.8269 |
| 17:0 | 0.17 ± 0.00 | 0.16 ± 0.02 | 0.33 ± 0.01 | 0.45 ± 0.01 | 0.32 ± 0.04 | 0.0357 |
| 18:0 | 10.73 ± 0.09 | 12.11 ± 0.47 | 12.56 ± 0.06 | 12.08 ± 0.23 | 18.39 ± 2.02 | 0.0267 |
| trans 18:1 | nd | nd | 0.20 ± 0.03 | 0.24 ± 0.07 | 0.03 ± 0.04 | 0.0379 |
| 18:1c9 | 15.87 ± 0.95 | 19.09 ± 1.20 | 16.05 ± 0.06 | 15.41 ± 0.53 | 12.10 ± 2.75 | 0.2525 |
| 18:1c11 | 2.70 ± 0.06 | 2.46 ± 0.07 | 1.94 ± 0.08 | 1.99 ± 0.12 | 1.81 ± 0.11 | 0.4285 |
| trans 18:2 | 0.10 ± 0.00 | 0.10 ± 0.01 | 0.28 ± 0.01 | 0.44 ± 0.09 | 0.14 ± 0.05 | 0.0437 |
| 18:2 ω-6 | 11.22 ± 0.30 | 11.67 ± 0.25 | 10.97 ± 0.12 | 11.61 ± 0.07 | 9.67 ± 2.05 | 0.5189 |
| 18:3 ω-6 | 0.05 ± 0.05 | 0.16 ± 0.02 | 0.18 ± 0.01 | 0.09 ± 0.09 | 0.02 ± 0.03 | 0.1602 |
| 18:3 ω-3 | 0.18 ± 0.04 | 0.18 ± 0.00 | 0.19 ± 0.03 | 0.26 ± 0.07 | 0.17 ± 0.04 | 0.3714 |
| 20:1c11 | 0.48 ± 0.02 | 0.39 ± 0.04 | 0.37 ± 0.01 | 0.40 ± 0.01 | 0.24 ± 0.03 | 0.0089 |
| 20:2 ω-6 | 0.55 ± 0.02 | 0.39 ± 0.07 | 0.35 ± 0.03 | 0.33 ± 0.02 | 0.35 ± 0.15 | 0.9857 |
| 20:3 ω-6 | 1.22 ± 0.01 | 1.14 ± 0.13 | 1.26 ± 0.10 | 1.26 ± 0.01 | 0.80 ± 0.08 | 0.0088 |
| trans 20:4 | 0.20 ± 0.03 | 0.24 ± 0.04 | 0.07 ± 0.07 | 0.68 ± 0.15 | 0.15 ± 0.05 | 0.0134 |
| 20:4 ω-6 | 11.53 ± 0.99 | 10.40 ± 0.95 | 15.81 ± 0.04 | 14.20 ± 0.09 | 10.07 ± 2.01 | 0.0463 |
| 20:5 ω-3 | 0.43 ± 0.03 | 0.42 ± 0.03 | 0.52 ± 0.03 | 0.53 ± 0.04 | 0.32 ± 0.07 | 0.0443 |
| 22:5 ω-3 | 0.89 ± 0.05 | 0.67 ± 0.04 | 0.76 ± 0.06 | 0.72 ± 0.05 | 0.87 ± 0.18 | 0.5865 |
| 22:6 ω-3 | 5.70 ± 0.08 | 4.84 ± 0.30 | 7.07 ± 0.03 | 7.60 ± 0.01 | 4.04 ± 0.74 | 0.0064 |
| Fatty Acid Families and Indices | Control (4 w) | Control (17 w) | Tumor-Bearing (4 w) | Tumor-Bearing (5 w) | Tumor-Bearing (17 w) | p Value |
|---|---|---|---|---|---|---|
| SFA | 46.99 ± 0.35 | 44.75 ± 0.93 | 42.16 ± 0.03 | 42.54 ± 1.07 | 57.86 ± 0.44 | <0.0001 |
| MUFA | 20.94 ± 1.20 | 25.06 ± 1.60 | 20.08 ± 0.08 | 19.53 ± 0.77 | 15.54 ± 3.34 | 0.0658 |
| PUFA | 32.07 ± 0.85 | 30.00 ± 0.48 | 37.09 ± 0.13 | 36.58 ± 0.01 | 26.60 ± 3.58 | 0.0190 |
| PUFA ω-6 | 24.86 ± 0.65 | 23.89 ± 0.77 | 28.56 ± 0.04 | 27.48 ± 0.04 | 21.21 ± 3.47 | 0.0960 |
| PUFA ω-3 | 7.21 ± 0.20 | 6.11 ± 0.29 | 8.54 ± 0.09 | 9.11 ± 0.03 | 5.39 ± 0.94 | 0.0054 |
| ω-6/ω-3 | 3.45 ± 0.01 | 3.93 ± 0.31 | 3.35 ± 0.03 | 3.02 ± 0.01 | 4.05 ± 1.02 | 0.5875 |
| SFA/MUFA | 2.25 ± 0.15 | 1.80 ± 0.15 | 2.10 ± 0.01 | 2.18 ± 0.14 | 3.89 ± 0.76 | 0.0281 |
| total trans | 0.30 ± 0.03 | 0.34 ± 0.05 | 0.55 ± 0.11 | 1.36 ± 0.32 | 0.32 ± 0.07 | 0.0074 |
| UI | 135.06 ± 3.01 | 128.63 ± 0.47 | 159.61 ± 0.53 | 156.82 ± 0.63 | 109.00 ± 10.19 | 0.0014 |
| PI | 114.39 ± 4.76 | 101.76 ± 1.50 | 142.52 ± 0.60 | 140.68 ± 0.24 | 92.10 ± 13.59 | 0.0053 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krokidis, M.G.; Louka, M.; Efthimiadou, E.K.; Zervou, S.-K.; Papadopoulos, K.; Hiskia, A.; Ferreri, C.; Chatgilialoglu, C. Membrane Lipidome Reorganization and Accumulation of Tissue DNA Lesions in Tumor-Bearing Mice: An Exploratory Study. Cancers 2019, 11, 480. https://doi.org/10.3390/cancers11040480
Krokidis MG, Louka M, Efthimiadou EK, Zervou S-K, Papadopoulos K, Hiskia A, Ferreri C, Chatgilialoglu C. Membrane Lipidome Reorganization and Accumulation of Tissue DNA Lesions in Tumor-Bearing Mice: An Exploratory Study. Cancers. 2019; 11(4):480. https://doi.org/10.3390/cancers11040480
Chicago/Turabian StyleKrokidis, Marios G., Maria Louka, Eleni K. Efthimiadou, Sevasti-Kiriaki Zervou, Kyriakos Papadopoulos, Anastasia Hiskia, Carla Ferreri, and Chryssostomos Chatgilialoglu. 2019. "Membrane Lipidome Reorganization and Accumulation of Tissue DNA Lesions in Tumor-Bearing Mice: An Exploratory Study" Cancers 11, no. 4: 480. https://doi.org/10.3390/cancers11040480
APA StyleKrokidis, M. G., Louka, M., Efthimiadou, E. K., Zervou, S.-K., Papadopoulos, K., Hiskia, A., Ferreri, C., & Chatgilialoglu, C. (2019). Membrane Lipidome Reorganization and Accumulation of Tissue DNA Lesions in Tumor-Bearing Mice: An Exploratory Study. Cancers, 11(4), 480. https://doi.org/10.3390/cancers11040480