Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients


Abstract
1. Introduction
2. Results
2.1. Study Participants
2.2. Patient, Tumour and Treatment Characteristics
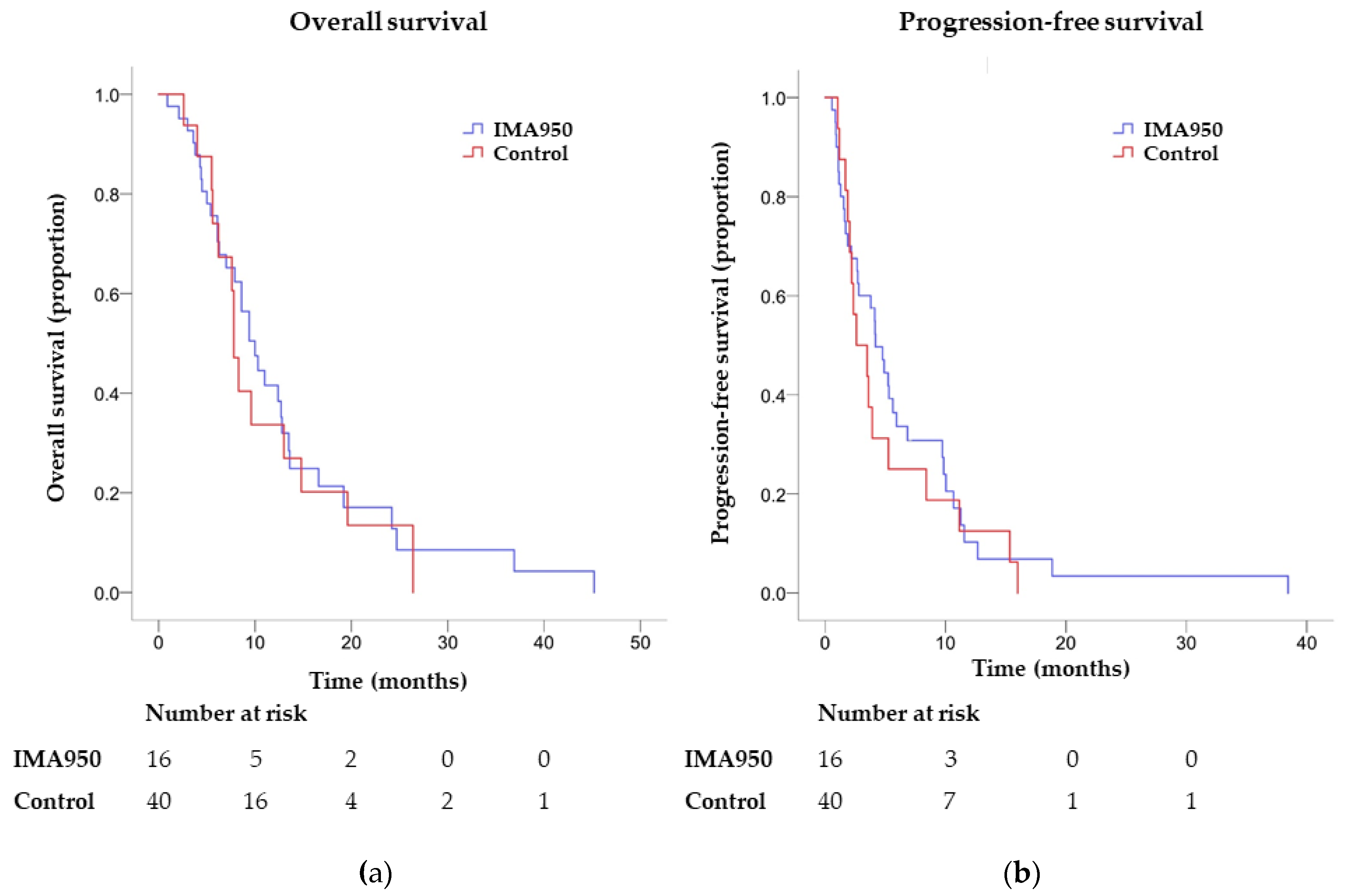
2.3. Endpoints
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. IMA950 Vaccine Treated Patients
4.3. Control Group
4.4. Data Collection
4.5. Endpoints
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Number of Recipients, n (%) | IMA950 n = 16 | Control n = 40 |
|---|---|---|
| Prior lines before BEV | 3 (18.8) | 15 (37.5) |
| Temozolomide | 1 (6.3) | 10 (25.0) |
| Lomustine | 1 (6.3) | 0 |
| Radiotherapy | 0 | 1 (2.5) |
| Sorafenib | 0 | 1 (2.5) |
| Pembrolizumab | 1 (6.3) | 0 |
| Rindopeptimut | 0 | 1 (2.5) |
| BGJ398 [42] | 0 | 1 (2.5) |
| GAPVAC-101 vaccination (glioma peptide vaccine plus poly-ICLC and GM-CSF) [43] | 0 | 1 (2.5) |
| Concomitant therapy to BEV at initiation | 4 (25.0) | 15 (35.0) |
| Lomustine | 2 (12.5) | 11 (27.5) |
| Irinotecan | 0 | 1 (2.5) |
| Temozolomide | 1 (6.3) | 1 (2.5) |
| Radiotherapy | 1 (6.3) | 0 |
| Buparlisib | 0 | 1 (2.5) |
| Subsequent lines after progression under BEV 1 | 12 (75.0) | 18 (45.0) |
| Lomustine | 7 (43.8) | 12 (30.0) |
| Irinotecan | 5 (31.3) | 10 (25.0) |
| Surgery | 2 (12.5) | 2 (5.0) |
| Radiotherapy | 2 (12.5) | 2 (5.0) |
| Nivolumab | 0 | 1 (2.5) |
| Tumour treating fields | 0 | 1 (2.5) |
| Etoposide | 0 | 1 (2.5) |
| Carboplatin | 1 (6.3) | 1 (2.5) |
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Wick, W.; Osswald, M.; Wick, A.; Winkler, F. Treatment of glioblastoma in adults. Ther. Adv. Neurol Disord. 2018, 11. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-Oncology 2019. [Google Scholar] [CrossRef]
- Rampling, R.; Peoples, S.; Mulholland, P.J.; James, A.; Al-Salihi, O.; Twelves, C.J.; McBain, C.; Jefferies, S.; Jackson, A.; Stewart, W.; et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2016, 22, 4776–4785. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012, 135, 1042–1054. [Google Scholar] [CrossRef]
- Dutoit, V.; Migliorini, D.; Dietrich, P.Y.; Walker, P.R. Immunotherapy of Malignant Tumors in the Brain: How Different from Other Sites? Front. Oncol. 2016, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Reardon, D.A. Concepts for Immunotherapies in Gliomas. Semin. Neurol. 2018, 38, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M.; van den Bent, M.; Stupp, R. Bevacizumab and recurrent malignant gliomas: A European perspective. J. Clin. Oncol. 2010, 28, e188–e189. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Roth, P.; Rushing, E.J.; Weller, J.; Andratschke, N.; Hofer, S.; Korol, D.; Regli, L.; Pangalu, A.; Pless, M.; et al. Bevacizumab may improve quality of life, but not overall survival in glioblastoma: An epidemiological study. Ann. Oncol. 2018, 29, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Hurwitz, H. Combinations of Bevacizumab with Cancer Immunotherapy. Cancer J. 2018, 24, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Fricke, I.; Mirza, N.; Dupont, J.; Lockhart, C.; Lee, J.H.; Sosman, J.A.; Gabrilovich, D.I. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin. Cancer Res. 2007, 13, 4840–4848. [Google Scholar] [CrossRef]
- Ohm, J.E.; Carbone, D.P. VEGF as a mediator of tumor-associated immunodeficiency. Immunol. Res. 2001, 23, 263–272. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar] [CrossRef]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Sahebjam, S.; Drury-Sibiga, A.; Etame, A.B.; Evernden, B.; Arrington, J.; Wicklund, M.; Jaglal, M.; Yu, M.; Tran, N.D.; Forsyth, P.; et al. ATIM-18. A phase I trial of hypofractionated stereotactic irradiation (HFSRT) with pembrolizumab and bevacizumab in patients with recurrent high grade glioma (NCT02313272). Neuro-Oncology 2017, 19, vi30. [Google Scholar] [CrossRef]
- Reardon, D.A.; Nayak, L.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; De Groot, J.F.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; Gaffey, S.C.; et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 2018, 36, 2006. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Nabors, L.B.; Kumthekar, P.; Badruddoja, M.A.; Fink, K.L.; Lieberman, F.S.; Phuphanich, S.; Dunbar, E.M.; Walbert, T.; Schiff, D.; et al. Phase 2 trial of SL-701 in relapsed/refractory (r/r) glioblastoma (GBM): Correlation of immune response with longer-term survival. J. Clin. Oncol. 2018, 36, 2058. [Google Scholar] [CrossRef]
- Bota, D.A.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.T.; Fu, B.D.; Hsu, F.P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: Interim results and correlations with CD4(+) T-lymphocyte counts. CNS Oncol. 2018, 7, CNS22. [Google Scholar] [CrossRef]
- Reardon, D.A.; Ashby, L.S.; Duic, J.P.; Mrugala, M.M.; Werner, A.; Turner, C.D.; Green, J.; Vitale, L.; Yellin, M.J. IMCT-08 ReACT: Long-term survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. Neuro-Oncology 2015, 17, v109. [Google Scholar] [CrossRef]
- Li, J.; Wang, M.; Won, M.; Shaw, E.G.; Coughlin, C.; Curran, W.J., Jr.; Mehta, M.P. Validation and simplification of the Radiation Therapy Oncology Group recursive partitioning analysis classification for glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, R.; MacLean, A.; Muzikansky, A.; Mukundan, S.; Wen, P.Y. Recurrent high-grade glioma treated with bevacizumab: Prognostic value of MGMT methylation, EGFR status and pretreatment MRI in determining response and survival. J. Neurooncol. 2013, 115, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Galanis, E.; DeGroot, J.F.; Cloughesy, T.F.; Wefel, J.S.; Lamborn, K.R.; Lassman, A.B.; Gilbert, M.R.; Sampson, J.H.; Wick, W.; et al. Clinical trial end points for high-grade glioma: The evolving landscape. Neuro-Oncology 2011, 13, 353–361. [Google Scholar] [CrossRef]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.; Beerepoot, L.V.; Hanse, M.C.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.; Dinjens, W.N.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Heiland, D.H.; Masalha, W.; Franco, P.; Machein, M.R.; Weyerbrock, A. Progression-free and overall survival in patients with recurrent Glioblastoma multiforme treated with last-line bevacizumab versus bevacizumab/lomustine. J. Neurooncol. 2016, 126, 567–575. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.N.; Urup, T.; Grunnet, K.; Toft, A.; Johansen, M.D.; Poulsen, S.H.; Christensen, I.J.; Muhic, A.; Poulsen, H.S. Toxicity and efficacy of lomustine and bevacizumab in recurrent glioblastoma patients. J. Neurooncol. 2018, 137, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Prawira, A.; Kaye, A.H.; Hovens, C.M. Tumour angiogenesis: Its mechanism and therapeutic implications in malignant gliomas. J. Clin. Neurosci. 2009, 16, 1119–1130. [Google Scholar] [CrossRef]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016, 1, e87059. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Macdonald, D.R.; Reardon, D.A.; Cloughesy, T.F.; Sorensen, A.G.; Galanis, E.; Degroot, J.; Wick, W.; Gilbert, M.R.; Lassman, A.B.; et al. Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. J. Clin. Oncol. 2010, 28, 1963–1972. [Google Scholar] [CrossRef]
- Chinot, O.L.; Macdonald, D.R.; Abrey, L.E.; Zahlmann, G.; Kerloëguen, Y.; Cloughesy, T.F. Response Assessment Criteria for Glioblastoma: Practical Adaptation and Implementation in Clinical Trials of Antiangiogenic Therapy. Curr. Neurol. Neurosci. Rep. 2013, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.P.; Hidalgo, M.; Schellens, J.H.M.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]



| Variable | IMA950 (n = 16) | Control (n = 40) | p-Value |
|---|---|---|---|
| Patient variables | |||
| Median age at BEV initiation (range) | 58 (24, 74) | 59.5 (33, 76) | 0.42 |
| Gender, n (%) | 0.47 | ||
| Male | 12 (75.0) | 26 (65.0) | |
| Female | 4 (25.0) | 14 (35.0) | |
| Tumour variables | |||
| Initial diagnosis, n (%) | 0.40 | ||
| Primary GBM | 13 (81.3) | 34 (85.0) | |
| Primary GBM + PNC | 1 (6.3) | 0 | |
| Granular cell astrocytoma | 0 | 1 (2.5) | |
| AA III | 2 (12.5) | 5 (12.5) | |
| MGMT gene promoter status, n (%) | 0.005 | ||
| Unmethylated | 14 (87.5) | 15 (37.5) | 0.001 |
| Methylated | 2 (12.5) | 7 (17.5) | 0.65 |
| Partially methylated | 0 | 1 (2.5) | 0.52 |
| Unknown | 0 | 17 (42.5) | 0.002 |
| IDH gene status, n (%) | 0.16 | ||
| Wild type | 15 (94.0) | 29 (72.5) | |
| Mutated | 1 (6.0) | 4 (10.0) | |
| Unknown | 0 | 7 (17.5) | |
| Management | |||
| Extent of resection, n (%) | 0.20 | ||
| Biopsy only | 2 (12.5) | 5 (12.5) | |
| Partial resection | 0 | 7 (17.5) | |
| Subtotal/total resection | 14 (87.5) | 28 70.0) | |
| Adjuvant TMZ | |||
| # recipients, n (%) | 16 (100) | 36 (90.0) | 0.19 |
| # cycles, median (CI) | 6.0 (1-) | 4 (0–12) | 0.35 |
| BEV | |||
| # BEV injections, median (CI) | 10.5 (3-) | 10.5 (1–73) | 0.77 |
| BEV regimen at initiation, n (%) | 0.37 | ||
| Monotherapy | 12 (75.0) | 25 (62.5) | |
| Combination therapy | 4 (25.0) | 15 (37.5) | |
| Line of treatment, n (%) | 0.17 | ||
| Second line | 14 (87.5) | 29 (70) | |
| > second line | 2 (12.5) | 12 (30) | |
| RPA groups, N (%) | |||
| III | 4 (25.0) | 5 (12.5) | 0.25 |
| IV | 7 (43.7) | 17 (42.5) | 0.73 |
| V | 5 (31.3) | 18 (45.0) | 0.35 |
| Outcome | IMA950 | Control | p-Value |
|---|---|---|---|
| From BEV initiation | |||
| OS, median (CI), months | 7.8 (6.9–8.7) | 10.0 (7.4–12.6) | 0.69 |
| PFS, median (CI), months | 2.6 (0.4–4.9) | 4.2 (2.8–5.5) | 0.50 |
| PFS-6, % | 25 | 30 | 0.71 |
| PFS-9, % | 18.8 | 22.5 | 0.76 |
| PFS-12, % | 12.5 | 7.5 | 0.55 |
| From initial diagnosis | |||
| OS, median (CI), months | 19.0 (18.0–20.0) | 25.0 (21.6–28.4) | 0.53 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boydell, E.; Marinari, E.; Migliorini, D.; Dietrich, P.-Y.; Patrikidou, A.; Dutoit, V. Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients. Cancers 2019, 11, 464. https://doi.org/10.3390/cancers11040464
Boydell E, Marinari E, Migliorini D, Dietrich P-Y, Patrikidou A, Dutoit V. Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients. Cancers. 2019; 11(4):464. https://doi.org/10.3390/cancers11040464
Chicago/Turabian StyleBoydell, Emma, Eliana Marinari, Denis Migliorini, Pierre-Yves Dietrich, Anna Patrikidou, and Valérie Dutoit. 2019. "Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients" Cancers 11, no. 4: 464. https://doi.org/10.3390/cancers11040464
APA StyleBoydell, E., Marinari, E., Migliorini, D., Dietrich, P.-Y., Patrikidou, A., & Dutoit, V. (2019). Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients. Cancers, 11(4), 464. https://doi.org/10.3390/cancers11040464