TRAILblazing Strategies for Cancer Treatment


{kind=link}
{kind=link}
Abstract
1. Introduction
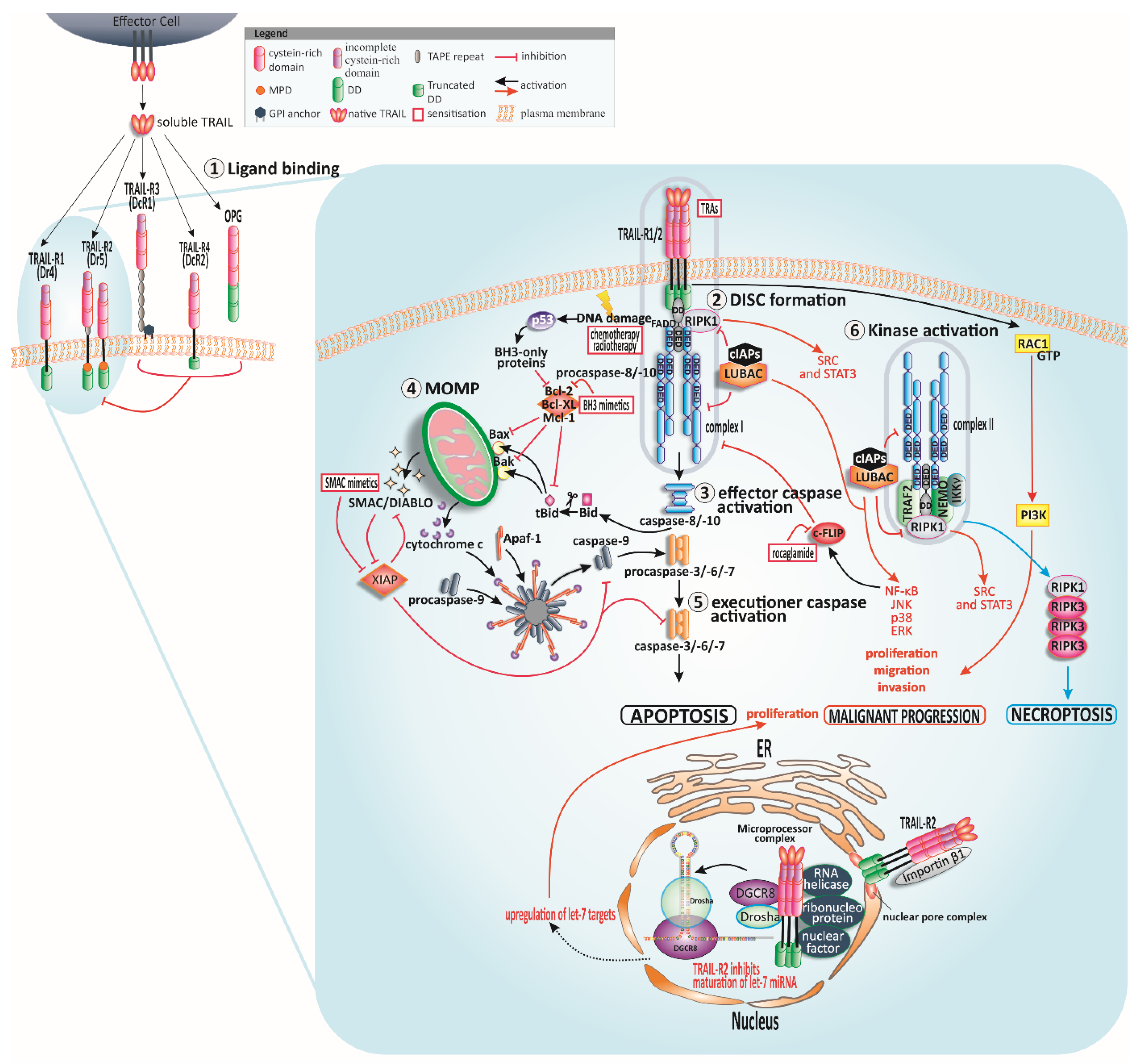
2. The TRAIL System
3. The Apoptotic ‘TRAIL’
3.1. TRAIL-Induced Extrinsic Apoptosis
3.2. TRAIL-Induced Cross-Signalling to Mitochondria
3.3. Checkpoints for TRAIL-Induced Apoptosis
4. On the TRAIL for Targeted Cancer Therapy
4.1. TRAs in Clinical Studies—Can Failure Still Lead to Success?

4.2. TRAIL Resistance Mechanisms in Cancer
4.2.1. TRAIL Receptors
4.2.2. The DISC
4.2.3. Bcl-2 Family
4.2.4. IAPs
4.2.5. Autophagy—The Self-Consuming TRAIL
4.2.6. Fractional Killing and Microenvironment
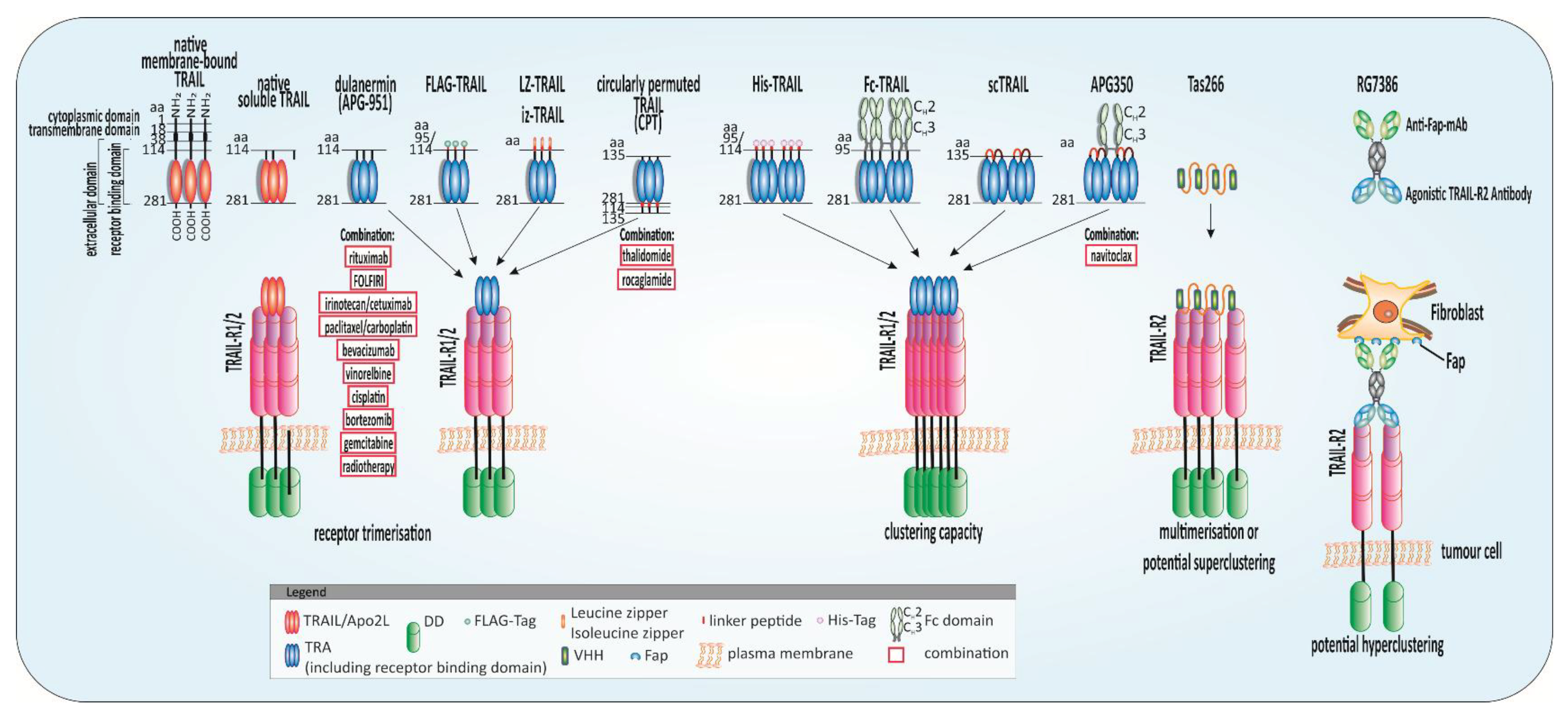
5. Strategies to Regain TRAIL Sensitivity—The Bench-to-Bedside TRAIL
5.1. Highly Potent TRAs
5.2. Chemo- and Radiotherapy Plus TRAIL
5.3. Inhibition of Anti-Apoptotic c-FLIP in Combination with TRAIL
5.4. BH3 and SMAC Mimetics Plus TRAIL
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| Apaf-1 | protease activating factor-1 |
| Apo2L | Apo-2 ligand |
| ATG | autophagy-related genes |
| Bak | Bcl-2 antagonist or killer |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell CLL/lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra-large |
| BH | Bcl-2 homology |
| Bid | BH3-only protein |
| BIR | baculovirus IAP repeat |
| CLL | chronic lymphocytic leukemia |
| CTC | circulating tumour cells |
| c-FLIP | cellular (FADD-like IL-1β-converting enzyme)-inhibitory protein |
| c-FLIPL | c-FLIP large |
| c-FLIPS | c-FLIP short |
| cIAP | cellular inhibitor of apoptosis protein |
| CPT | circularly permuted TRAIL |
| DcR | decoy receptor |
| DD | death domain |
| DED | death effector domains |
| DISC | death-inducing signalling complex |
| DLT | dose-limiting toxicity |
| DR | death receptor |
| ERK | extracellular signal-regulated kinases |
| FADD | Fas-associated death domain |
| FAP | fibroblast-activation protein |
| Fc | fragment crystallizable region |
| FDA | United States Food and Drug Administration |
| FLICE | FADD-like IL-1β-converting enzyme |
| FOLFIRI | folinic acid, fluorouracil, irinotecan |
| GALNT14 | polypeptide N-acetylgalactosaminyltransferase 14 |
| GPI | glycosylphosphatidylinositol |
| HCC | hepatocellular carcinoma |
| HSA | human serum albumin |
| HTS | high-throughput screenings |
| IgG | Immunoglobulin G |
| IKK | inhibitor of κB kinase |
| IL | interleukin |
| iz | isoleucin |
| IκB | inhibitor of κB |
| JNK | c-Jun N-terminal kinases |
| KRAS | Kirsten rat sarcoma |
| LUBAC | linear ubiquitin chain assembly complex |
| LZ | leucine zipper |
| MAPK | mitogen-activated protein kinase |
| Mcl-1 | myeloid leukaemia cell differentiation protein |
| miRNA | microRNA |
| MOM | mitochondrial outer membrane |
| MOMP | mitochondrial outer membrane permeabilization |
| MPD | membrane-proximal domain |
| MTD | maximum tolerated dose |
| mTOR | mechanistic target of rapamycin |
| NEMO | NF-κB essential modifier |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NIAP | neuronal apoptosis inhibitory protein |
| NSCLC | non-small-cell lung cancer |
| OPG | osteoprotegerin |
| ORR | objective response rate |
| OS | overall survival |
| PFS | progression-free survival |
| pI | isoelectric point |
| PI3K | tyrosine kinase Src and phosphoinositide 3-kinase |
| PUMA | p53 upregulated modulator of apoptosis |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| rhTRAIL | recombinant human TRAIL |
| RIPK | receptor-interacting protein kinase |
| RRMM | relapsed refractory multiple myeloma |
| scTRAIL | single-chain TRAIL |
| SMAC/DIABLO | second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low pI |
| SR | superfamily |
| tBID | truncated BH3-only protein |
| TD | thalidomide/dexamethasone |
| TNF | tumor necrosis factor |
| TNF-SF | TNF-superfamily |
| TRAF2 | TNF receptor-associated factor-2 |
| TRAE | treatment-related adverse events |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TRAIL-R | TRAIL receptor |
| TRA | TRAIL receptor agonist |
| Ts-IAP | testis-specific IAP |
| USP8 | ubiquitin-specific peptidase 8 |
| VHH | high-affinity heavy chain domain |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science (N. Y.) 1997, 277, 815–818. [Google Scholar] [CrossRef]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; de Vos, A.M. Triggering cell death: The crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- Mongkolsapaya, J.; Grimes, J.M.; Chen, N.; Xu, X.N.; Stuart, D.I.; Jones, E.Y.; Screaton, G.R. Structure of the TRAIL-DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat. Struct. Biol. 1999, 6, 1048–1053. [Google Scholar]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar] [CrossRef]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef]
- De Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science (N. Y.) 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science (N. Y.) 1997, 277, 818–821. [Google Scholar] [CrossRef]
- Marsters, S.A.; Sheridan, J.P.; Pitti, R.M.; Huang, A.; Skubatch, M.; Baldwin, D.; Yuan, J.; Gurney, A.; Goddard, A.D.; Godowski, P.; et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr. Biol. 1997, 7, 1003–1006. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Takahashi, N.; Udagawa, N.; Suda, T. A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem. Biophys. Res. Commun. 1999, 256, 449–455. [Google Scholar] [CrossRef]
- Truneh, A.; Sharma, S.; Silverman, C.; Khandekar, S.; Reddy, M.P.; Deen, K.C.; McLaughlin, M.M.; Srinivasula, S.M.; Livi, G.P.; Marshall, L.A.; et al. Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J. Biol. Chem. 2000, 275, 23319–23325. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; Noack, A.; Adam, D.; Tchikov, V.; Bertsch, U.; Roder, C.; Schutze, S.; Wajant, H.; Kalthoff, H.; Trauzold, A. TRAIL signaling is mediated by DR4 in pancreatic tumor cells despite the expression of functional DR5. J. Mol. Med. 2010, 88, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Stadel, D.; Mohr, A.; Ref, C.; MacFarlane, M.; Zhou, S.; Humphreys, R.; Bachem, M.; Cohen, G.; Moller, P.; Zwacka, R.M.; et al. TRAIL-induced apoptosis is preferentially mediated via TRAIL receptor 1 in pancreatic carcinoma cells and profoundly enhanced by XIAP inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5734–5749. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.; Inoue, S.; Kohlhaas, S.L.; Majid, A.; Harper, N.; Kennedy, D.B.; Dyer, M.J.; Cohen, G.M. Chronic lymphocytic leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death Differ. 2005, 12, 773–782. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.; Kohlhaas, S.L.; Sutcliffe, M.J.; Dyer, M.J.; Cohen, G.M. TRAIL receptor-selective mutants signal to apoptosis via TRAIL-R1 in primary lymphoid malignancies. Cancer Res. 2005, 65, 11265–11270. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999, 59, 2770–2775. [Google Scholar]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 2006, 281, 13964–13971. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Wang, T.T.; Jeng, J. Coordinated regulation of two TRAIL-R2/KILLER/DR5 mRNA isoforms by DNA damaging agents, serum and 17beta-estradiol in human breast cancer cells. Breast Cancer Res. Treat. 2000, 61, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Wallace-Brodeur, R.R.; Lowe, S.W. Clinical implications of p53 mutations. Cell. Mol. Life Sci. 1999, 55, 64–75. [Google Scholar] [CrossRef]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging 2012, 4, 330–349. [Google Scholar] [CrossRef]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Maecker, H.; Sharp, D.; Lawrence, D.; Renz, M.; Vucic, D.; Ashkenazi, A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 2005, 280, 40599–40608. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE Signal Transduct. Knowl. Environ. 2006, 2006, re13. [Google Scholar] [CrossRef] [PubMed]
- Karl, I.; Jossberger-Werner, M.; Schmidt, N.; Horn, S.; Goebeler, M.; Leverkus, M.; Wajant, H.; Giner, T. TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell Death Dis. 2014, 5, e1444. [Google Scholar] [CrossRef] [PubMed]
- Lafont, E.; Kantari-Mimoun, C.; Draber, P.; De Miguel, D.; Hartwig, T.; Reichert, M.; Kupka, S.; Shimizu, Y.; Taraborrelli, L.; Spit, M.; et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017, 36, 1147–1166. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Yuvaraj, S.; Peppelenbosch, M.P.; Wurdinger, T.; Dekker, H.; Joore, J.; van Dijk, E.; Quax, W.J.; Peters, G.J.; de Jong, S.; et al. Kinome profiling of non-canonical TRAIL signaling reveals RIP1-Src-STAT3-dependent invasion in resistant non-small cell lung cancer cells. J. Cell Sci. 2012, 125 Pt 19, 4651–4661. [Google Scholar] [CrossRef]
- Von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Haselmann, V.; Kurz, A.; Bertsch, U.; Hubner, S.; Olempska-Muller, M.; Fritsch, J.; Hasler, R.; Pickl, A.; Fritsche, H.; Annewanter, F.; et al. Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumor cells. Gastroenterology 2014, 146, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Lawrence, D.A.; Tinel, A.; LeBlanc, H.; Virmani, A.; Schow, P.; Gazdar, A.; Blenis, J.; Arnott, D.; Ashkenazi, A. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J. Biol. Chem. 2001, 276, 46639–46646. [Google Scholar] [CrossRef] [PubMed]
- Sprick, M.R.; Rieser, E.; Stahl, H.; Grosse-Wilde, A.; Weigand, M.A.; Walczak, H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002, 21, 4520–4530. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; Macfarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef]
- Schleich, K.; Buchbinder, J.H.; Pietkiewicz, S.; Kahne, T.; Warnken, U.; Ozturk, S.; Schnolzer, M.; Naumann, M.; Krammer, P.H.; Lavrik, I.N. Molecular architecture of the DED chains at the DISC: Regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016, 23, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw-Makepeace, J.; Huston, G.; Fortner, K.A.; Russell, J.Q.; Holoch, D.; Swain, S.; Budd, R.C. C-FLIP(S) reduces activation of caspase and NF-kappaB pathways and decreases T cell survival. Eur. J. Immunol. 2008, 38, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Arhoma, A.; Chantry, A.D.; Haywood-Small, S.L.; Cross, N.A. SAHA-induced TRAIL-sensitisation of Multiple Myeloma cells is enhanced in 3D cell culture. Exp. Cell Res. 2017, 360, 226–235. [Google Scholar] [CrossRef]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Rasper, D.M.; Vaillancourt, J.P.; Hadano, S.; Houtzager, V.M.; Seiden, I.; Keen, S.L.; Tawa, P.; Xanthoudakis, S.; Nasir, J.; Martindale, D.; et al. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998, 5, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.K.; Wang, L.; Zheng, L.; Wan, F.; Ahmed, M.; Lenardo, M.J.; Wu, H. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol. Cell 2005, 20, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.; Schmitz, I.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 2001, 276, 20633–20640. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, S.; Schleich, K.; Lavrik, I.N. Cellular FLICE-like inhibitory proteins (c-FLIPs): Fine-tuners of life and death decisions. Exp. Cell Res. 2012, 318, 1324–1331. [Google Scholar] [CrossRef]
- Fricker, N.; Beaudouin, J.; Richter, P.; Eils, R.; Krammer, P.H.; Lavrik, I.N. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J. Cell Biol. 2010, 190, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Thome, M.; Schneider, P.; Holler, N.; Tschopp, J.; Nicholson, D.W.; Briand, C.; Grutter, M.G. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 2002, 277, 45162–45171. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Jeffrey, P.D.; Shi, Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc. Natl. Acad. Sci. USA 2009, 106, 8169–8174. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Deis, C.; Denault, J.B.; Sutherlin, D.P.; Salvesen, G.S. Activation of caspases-8 and -10 by FLIP (L). Biochem. J. 2004, 382 Pt 2, 651–657. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lin, Y.; Wu, X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002, 16, 33–45. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.; Lawrence, D.; Varfolomeev, E.; Totpal, K.; Morlan, J.; Schow, P.; Fong, S.; Schwall, R.; Sinicropi, D.; Ashkenazi, A. Tumor-cell resistance to death receptor–induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat. Med. 2002, 8, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Ozören, N.; El-Deiry, W.S. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia (N. Y.) 2002, 4, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Grabow, S.; Gray, D.; McKenzie, M.D.; Nachbur, U.; Huang, D.C.S.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J.; et al. XIAP acts as a switch between type I and type II FAS-induced apoptosis signalling. Nature 2009, 460, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [CrossRef]
- Holcik, M.; Korneluk, R.G. XIAP, the guardian angel. Nat. Rev. Mol. Cell Biol. 2001, 2, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Bratton, S.B.; Walker, G.; Srinivasula, S.M.; Sun, X.M.; Butterworth, M.; Alnemri, E.S.; Cohen, G.M. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 2001, 20, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Antonsson, B.; Montessuit, S.; Sanchez, B.; Martinou, J.C. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 2001, 276, 11615–11623. [Google Scholar] [CrossRef] [PubMed]
- Dewson, G.; Kratina, T.; Czabotar, P.; Day, C.L.; Adams, J.M.; Kluck, R.M. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell 2009, 36, 696–703. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Fraser, C.; Muthalagu, N.; Bhola, P.D.; Chang, W.; McBrayer, S.K.; Cantlon, A.; Fisch, S.; Golomb-Mello, G.; Ryan, J.A.; et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 2017, 31, 142–156. [Google Scholar] [CrossRef]
- Zhang, X.D.; Nguyen, T.; Thomas, W.D.; Sanders, J.E.; Hersey, P. Mechanisms of resistance of normal cells to TRAIL induced apoptosis vary between different cell types. FEBS Lett. 2000, 482, 193–199. [Google Scholar]
- Singh, H.D.; Otano, I.; Rombouts, K.; Singh, K.P.; Peppa, D.; Gill, U.S.; Böttcher, K.; Kennedy, P.T.F.; Oben, J.; Pinzani, M.; et al. TRAIL regulatory receptors constrain human hepatic stellate cell apoptosis. Sci. Rep. 2017, 7, 5514. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.; Lalaoui, N.; Morizot, A.; Schneider, P.; Solary, E.; Micheau, O. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol. Cell. Biol. 2006, 26, 7046–7055. [Google Scholar] [CrossRef]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Halpin-McCormick, A.; Sessler, T.; Samali, A.; Szegezdi, E. Resistance to TRAIL in non-transformed cells is due to multiple redundant pathways. Cell Death Dis. 2013, 4, e702. [Google Scholar] [CrossRef]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.L.; von Karstedt, S.; Hillenbrand, A.; Henne-Bruns, D.; Knippschild, U.; Trauzold, A.; Lemke, J. Should We Keep Walking along the Trail for Pancreatic Cancer Treatment? Revisiting TNF-Related Apoptosis-Inducing Ligand for Anticancer Therapy. Cancers 2018, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Zhou, S.; Diaz, L.A., Jr.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739–751. [Google Scholar] [CrossRef]
- Soria, J.C.; Smit, E.; Khayat, D.; Besse, B.; Yang, X.; Hsu, C.P.; Reese, D.; Wiezorek, J.; Blackhall, F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 1527–1533. [Google Scholar] [CrossRef]
- Yee, L.; Fanale, M.; Dimick, K.; Calvert, S.; Robins, C.; Ling, I.J.; Novotny, W.; Ashkenazi, A., III. A phase IB safety and pharmacokinetic (PK) study of recombinant human Apo2L/TRAIL in combination with rituximab in patients with low-grade non-Hodgkin lymphoma. J. Clin. Oncol. 2007, 25 (Suppl. 18), 8078. [Google Scholar]
- Yee, L.; Burris, H.A.; Kozloff, M.; Wainberg, Z.; Pao, M.; Skettino, S.; Novotny, W.; Durbin, B.; Weston, J.; Hurwitz, H. Phase Ib study of recombinant human Apo2L/TRAIL plus irinotecan and cetuximab or FOLFIRI in metastatic colorectal cancer (mCRC) patients (pts): Preliminary results. J. Clin. Oncol. 2009, 27 (Suppl. 15), 4129. [Google Scholar]
- Kasubhai, S.M.; Bendell, J.C.; Kozloff, M.; Kapp, A.V.; Ashkenazi, A.; Royer-Joo, S.; Portera, C.C. Phase Ib study of dulanermin combined with FOLFIRI (with or without bevacizumab [BV]) in previously treated patients (Pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2012, 30 (Suppl. 15), 3543. [Google Scholar]
- Wainberg, Z.A.; Messersmith, W.A.; Peddi, P.F.; Kapp, A.V.; Ashkenazi, A.; Royer-Joo, S.; Portera, C.C.; Kozloff, M.F. A phase 1B study of dulanermin in combination with modified FOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Clin. Color. Cancer 2013, 12, 248–254. [Google Scholar] [CrossRef]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Cheah, C.Y.; Belada, D.; Fanale, M.A.; Janikova, A.; Czucman, M.S.; Flinn, I.W.; Kapp, A.V.; Ashkenazi, A.; Kelley, S.; Bray, G.L.; et al. Dulanermin with rituximab in patients with relapsed indolent B-cell lymphoma: An open-label phase 1b/2 randomised study. Lancet Haematol. 2015, 2, e166–e174. [Google Scholar] [CrossRef]
- Soria, J.C.; Mark, Z.; Zatloukal, P.; Szima, B.; Albert, I.; Juhasz, E.; Pujol, J.L.; Kozielski, J.; Baker, N.; Smethurst, D.; et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4442–4451. [Google Scholar] [CrossRef]
- Quintavalle, C.; Condorelli, G. Dulanermin in cancer therapy: Still much to do. Transl. Lung Cancer Res. 2012, 1, 158–159. [Google Scholar] [PubMed]
- Pan, Y.; Xu, R.; Peach, M.; Huang, C.P.; Branstetter, D.; Novotny, W.; Herbst, R.S.; Eckhardt, S.G.; Holland, P.M. Evaluation of pharmacodynamic biomarkers in a Phase 1a trial of dulanermin (rhApo2L/TRAIL) in patients with advanced tumours. Br. J. Cancer 2011, 105, 1830–1838. [Google Scholar] [CrossRef]
- Faller, B.A.; Pandit, T.N. Safety and efficacy of vinorelbine in the treatment of non-small cell lung cancer. Clin. Med. Insights Oncol. 2011, 5, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ceja, K.A.; Chirino, Y.I. Current FDA-approved treatments for non-small cell lung cancer and potential biomarkers for its detection. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 90, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Shi, M.; Jie, F.; Bai, Y.; Shen, P.; Yu, Z.; Wang, X.; Huang, C.; Tao, M.; Wang, Z.; et al. Phase III study of dulanermin (recombinant human tumor necrosis factor-related apoptosis-inducing ligand/Apo2 ligand) combined with vinorelbine and cisplatin in patients with advanced non-small-cell lung cancer. Investig. New Drugs 2018, 36, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Lefko, P.J.; Graves, J.D.; Zoog, S.J.; Pan, Y.; Wall, J.; Branstetter, D.G.; Moriguchi, J.; Coxon, A.; Huard, J.N.; Xu, R.; et al. Conatumumab, a fully human agonist antibody to death receptor 5, induces apoptosis via caspase activation in multiple tumor types. Cancer Biol. Ther. 2010, 9, 618–631. [Google Scholar] [CrossRef]
- Kang, Z.; Chen, J.J.; Yu, Y.; Li, B.; Sun, S.Y.; Zhang, B.; Cao, L. Drozitumab, a human antibody to death receptor 5, has potent antitumor activity against rhabdomyosarcoma with the expression of caspase-8 predictive of response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 3181–3192. [Google Scholar] [CrossRef] [PubMed]
- Rosevear, H.M.; Lightfoot, A.J.; Griffith, T.S. Conatumumab, a fully human mAb against death receptor 5 for the treatment of cancer. Curr. Opin. Investig. Drugs 2010, 11, 688–698. [Google Scholar]
- Sharma, S.; de Vries, E.G.; Infante, J.R.; Oldenhuis, C.N.; Gietema, J.A.; Yang, L.; Bilic, S.; Parker, K.; Goldbrunner, M.; Scott, J.W.; et al. Safety, pharmacokinetics, and pharmacodynamics of the DR5 antibody LBY135 alone and in combination with capecitabine in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 135–144. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Shah, J.; Wood, T.; Posey, J.; Carlisle, R.; Copigneaux, C.; Luo, F.; Wojtowicz-Praga, S.; Percent, I.; Saleh, M. Phase I Trial of Weekly Tigatuzumab, an Agonistic Humanized Monoclonal Antibody Targeting Death Receptor 5 (DR5). Cancer Biother. Radiopharm. 2010, 25, 13–19. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Infante, J.R.; Waterhouse, D.; Wong, L.; Vickers, S.; Arrowsmith, E.; He, A.R.; Hart, L.; Trent, D.; Wade, J.; et al. Phase 2, multicenter, open-label study of tigatuzumab (CS-1008), a humanized monoclonal antibody targeting death receptor 5, in combination with gemcitabine in chemotherapy-naive patients with unresectable or metastatic pancreatic cancer. Cancer Med. 2013, 2, 925–932. [Google Scholar] [CrossRef]
- Merchant, M.S.; Geller, J.I.; Baird, K.; Chou, A.J.; Galli, S.; Charles, A.; Amaoko, M.; Rhee, E.H.; Price, A.; Wexler, L.H.; et al. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 4141–4147. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Fakih, M.; Schwartzberg, L.; Cohn, A.L.; Yee, L.; Dreisbach, L.; Kozloff, M.F.; Hei, Y.J.; Galimi, F.; Pan, Y.; et al. TRAIL receptor agonist conatumumab with modified FOLFOX6 plus bevacizumab for first-line treatment of metastatic colorectal cancer: A randomized phase 1b/2 trial. Cancer 2013, 119, 4290–4298. [Google Scholar] [CrossRef]
- Gillies, R.J.; Schornack, P.A.; Secomb, T.W.; Raghunand, N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia (N. Y.) 1999, 1, 197–207. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011, 19, 101–113. [Google Scholar] [CrossRef]
- Dhein, J.; Daniel, P.T.; Trauth, B.C.; Oehm, A.; Möller, P.; Krammer, P.H. Induction of apoptosis by monoclonal antibody anti-APO-1 class switch variants is dependent on cross-linking of APO-1 cell surface antigens. J. Immunol. 1992, 149, 3166–3173. [Google Scholar] [PubMed]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070. [Google Scholar] [CrossRef]
- Huet, H.A.; Growney, J.D.; Johnson, J.A.; Li, J.; Bilic, S.; Ostrom, L.; Zafari, M.; Kowal, C.; Yang, G.; Royo, A.; et al. Multivalent nanobodies targeting death receptor 5 elicit superior tumor cell killing through efficient caspase induction. mAbs 2014, 6, 1560–1570. [Google Scholar] [CrossRef]
- Van Audenhove, I.; Gettemans, J. Nanobodies as Versatile Tools to Understand, Diagnose, Visualize and Treat Cancer. EBioMedicine 2016, 8, 40–48. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Isaacs, R.; Bilic, S.; Kentsch, K.; Huet, H.A.; Hofmann, M.; Rasco, D.; Kundamal, N.; Tang, Z.; Cooksey, J.; et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody(R) targeting the DR5 receptor. Cancer Chemother. Pharmacol. 2015, 75, 887–895. [Google Scholar] [CrossRef]
- Ganten, T.M.; Koschny, R.; Sykora, J.; Schulze-Bergkamen, H.; Buchler, P.; Haas, T.L.; Schader, M.B.; Untergasser, A.; Stremmel, W.; Walczak, H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Wang, A.P.; Yang, S.F. Antitumor activity of a novel recombinant mutant human tumor necrosis factor-related apoptosis-inducing ligand. Acta Pharmacol. Sin. 2005, 26, 1373–1381. [Google Scholar] [CrossRef]
- Wang, H.; Davis, J.S.; Wu, X. Immunoglobulin Fc domain fusion to TRAIL significantly prolongs its plasma half-life and enhances its antitumor activity. Mol. Cancer Ther. 2014, 13, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Munkel, S.; Krippner-Heidenreich, A.; Grunwald, I.; Wels, W.S.; Wajant, H.; Pfizenmaier, K.; Gerspach, J. Potent antitumoral activity of TRAIL through generation of tumor-targeted single-chain fusion proteins. Cell Death Dis. 2010, 1, e68. [Google Scholar] [CrossRef] [PubMed]
- Gieffers, C.; Kluge, M.; Merz, C.; Sykora, J.; Thiemann, M.; Schaal, R.; Fischer, C.; Branschadel, M.; Abhari, B.A.; Hohenberger, P.; et al. APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcgamma receptors. Mol. Cancer Ther. 2013, 12, 2735–2747. [Google Scholar] [CrossRef] [PubMed]
- Brunker, P.; Wartha, K.; Friess, T.; Grau-Richards, S.; Waldhauer, I.; Koller, C.F.; Weiser, B.; Majety, M.; Runza, V.; Niu, H.; et al. RG7386, a Novel Tetravalent FAP-DR5 Antibody, Effectively Triggers FAP-Dependent, Avidity-Driven DR5 Hyperclustering and Tumor Cell Apoptosis. Mol. Cancer Ther. 2016, 15, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Qiu, L.; Hou, J.; Zhang, X.; Ke, X.; Wang, Z.; Zhou, F.; Yang, S.; Zhao, Y.; Leng, Y.; et al. Phase Ib Study of Recombinant Circularly Permuted TRAIL (CPT) in Relapsed or Refractory multiple Myeloma Patients. Blood 2012, 120, 1857. [Google Scholar]
- Leng, Y.; Qiu, L.; Hou, J.; Zhao, Y.; Zhang, X.; Yang, S.; Xi, H.; Huang, Z.; Pan, L.; Chen, W. Phase II open-label study of recombinant circularly permuted TRAIL as a single-agent treatment for relapsed or refractory multiple myeloma. Chin. J. Cancer 2016, 35, 86. [Google Scholar] [CrossRef] [PubMed]
- Ridings, J.E. The thalidomide disaster, lessons from the past. Methods Mol. Biol. 2013, 947, 575–586. [Google Scholar]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef]
- Geng, C.; Hou, J.; Zhao, Y.; Ke, X.; Wang, Z.; Qiu, L.; Xi, H.; Wang, F.; Wei, N.; Liu, Y.; et al. A multicenter, open-label phase II study of recombinant CPT (Circularly Permuted TRAIL) plus thalidomide in patients with relapsed and refractory multiple myeloma. Am. J. Hematol. 2014, 89, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Hou, J.; Jin, J.; Zhang, M.; Ke, X.; Jiang, B.; Pan, L.; Yang, L.; Zhou, F.; Wang, J.; et al. Circularly permuted TRAIL plus thalidomide and dexamethasone versus thalidomide and dexamethasone for relapsed/refractory multiple myeloma: A phase 2 study. Cancer Chemother. Pharmacol. 2017, 79, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Dobos, J.; Kenessey, I.; Timar, J.; Ladanyi, A. Glucocorticoid receptor expression and antiproliferative effect of dexamethasone on human melanoma cells. Pathol. Oncol. Res. 2011, 17, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.K.; Smith, M.L.; Solomon, L.R.; Zhang, H.; Xue, J.C.; Xiao, Y.; Cheng, D.; Buchanan, G.; Morgan-Lappe, S.; Phillips, D.C. Abbv-621 Is a Novel and Potent TRAIL Receptor Agonist Fusion Protein That Induces Apoptosis Alone and in Combination with Navitoclax and Venetoclax in Hematological Tumors. Blood 2017, 130 (Suppl. 1), 2812. [Google Scholar]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, L.Y.; Anderson, C.K.; Camidge, R.; Behbakht, K.; Thorburn, A.; Ford, H.L. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene 2013, 32, 1341–1350. [Google Scholar] [CrossRef]
- Thorburn, A.; Behbakht, K.; Ford, H. TRAIL receptor-targeted therapeutics: Resistance mechanisms and strategies to avoid them. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2008, 11, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hopkins-Donaldson, S.; Bodmer, J.L.; Bourloud, K.B.; Brognara, C.B.; Tschopp, J.; Gross, N. Loss of caspase-8 expression in neuroblastoma is related to malignancy and resistance to TRAIL-induced apoptosis. Med. Pediatr. Oncol. 2000, 35, 608–611. [Google Scholar] [CrossRef]
- Eggert, A.; Grotzer, M.A.; Zuzak, T.J.; Wiewrodt, B.R.; Ho, R.; Ikegaki, N.; Brodeur, G.M. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001, 61, 1314–1319. [Google Scholar] [PubMed]
- Zhang, L.; Zhu, H.; Teraishi, F.; Davis, J.J.; Guo, W.; Fan, Z.; Fang, B. Accelerated degradation of caspase-8 protein correlates with TRAIL resistance in a DLD1 human colon cancer cell line. Neoplasia (N. Y.) 2005, 7, 594–602. [Google Scholar] [CrossRef]
- Lee, S.H.; Shin, M.S.; Kim, H.S.; Lee, H.K.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Han, S.Y.; Park, J.Y.; Oh, R.R.; et al. Somatic mutations of TRAIL-receptor 1 and TRAIL-receptor 2 genes in non-Hodgkin’s lymphoma. Oncogene 2001, 20, 399–403. [Google Scholar] [CrossRef]
- Fisher, M.J.; Virmani, A.K.; Wu, L.; Aplenc, R.; Harper, J.C.; Powell, S.M.; Rebbeck, T.R.; Sidransky, D.; Gazdar, A.F.; El-Deiry, W.S. Nucleotide substitution in the ectodomain of trail receptor DR4 is associated with lung cancer and head and neck cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1688–1697. [Google Scholar]
- Shin, M.S.; Kim, H.S.; Lee, S.H.; Park, W.S.; Kim, S.Y.; Park, J.Y.; Lee, J.H.; Lee, S.K.; Lee, S.N.; Jung, S.S.; et al. Mutations of tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1) and receptor 2 (TRAIL-R2) genes in metastatic breast cancers. Cancer Res. 2001, 61, 4942–4946. [Google Scholar] [PubMed]
- Sanlioglu, A.D.; Dirice, E.; Aydin, C.; Erin, N.; Koksoy, S.; Sanlioglu, S. Surface TRAIL decoy receptor-4 expression is correlated with TRAIL resistance in MCF7 breast cancer cells. BMC Cancer 2005, 5, 54. [Google Scholar] [CrossRef]
- Neumann, S.; Hasenauer, J.; Pollak, N.; Scheurich, P. Dominant negative effects of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 4 on TRAIL receptor 1 signaling by formation of heteromeric complexes. J. Biol. Chem. 2014, 289, 16576–16587. [Google Scholar] [CrossRef]
- O’Leary, L.; van der Sloot, A.M.; Reis, C.R.; Deegan, S.; Ryan, A.E.; Dhami, S.P.; Murillo, L.S.; Cool, R.H.; Correa de Sampaio, P.; Thompson, K.; et al. Decoy receptors block TRAIL sensitivity at a supracellular level: The role of stromal cells in controlling tumour TRAIL sensitivity. Oncogene 2016, 35, 1261–1270. [Google Scholar] [CrossRef]
- Trauzold, A.; Siegmund, D.; Schniewind, B.; Sipos, B.; Egberts, J.; Zorenkov, D.; Emme, D.; Roder, C.; Kalthoff, H.; Wajant, H. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006, 25, 7434–7439. [Google Scholar] [CrossRef]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e735. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O. Regulation of TNF-Related Apoptosis-Inducing Ligand Signaling by Glycosylation. Int. J. Mol. Sci. 2018, 19, 715. [Google Scholar] [CrossRef]
- Dufour, F.; Rattier, T.; Shirley, S.; Picarda, G.; Constantinescu, A.A.; Morle, A.; Zakaria, A.B.; Marcion, G.; Causse, S.; Szegezdi, E.; et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 2017, 24, 500–510. [Google Scholar] [CrossRef]
- Yoshida, T.; Shiraishi, T.; Horinaka, M.; Wakada, M.; Sakai, T. Glycosylation modulates TRAIL-R1/death receptor 4 protein: Different regulations of two pro-apoptotic receptors for TRAIL by tunicamycin. Oncol. Rep. 2007, 18, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Williams, J.L.; Greenwood, S.L.; Baker, P.N.; Aplin, J.D.; Crocker, I.P. A placental protective role for trophoblast-derived TNF-related apoptosis-inducing ligand (TRAIL). Placenta 2009, 30, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, U.; Röder, C.; Kalthoff, H.; Trauzold, A. Compartmentalization of TNF-related apoptosis-inducing ligand (TRAIL) death receptor functions: Emerging role of nuclear TRAIL-R2. Cell Death Dis. 2014, 5, e1390. [Google Scholar] [CrossRef] [PubMed]
- Kuang, A.A.; Diehl, G.E.; Zhang, J.; Winoto, A. FADD is required for DR4- and DR5-mediated apoptosis: Lack of trail-induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J. Biol. Chem. 2000, 275, 25065–25068. [Google Scholar] [CrossRef]
- Zang, F.; Wei, X.; Leng, X.; Yu, M.; Sun, B. C-FLIP (L) contributes to TRAIL resistance in HER2-positive breast cancer. Biochem. Biophys. Res. Commun. 2014, 450, 267–273. [Google Scholar] [CrossRef]
- Kaminskyy, V.O.; Surova, O.V.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Andera, L.; Zhivotovsky, B. Upregulation of c-FLIP-short in response to TRAIL promotes survival of NSCLC cells, which could be suppressed by inhibition of Ca2+/calmodulin signaling. Cell Death Dis. 2013, 4, e522. [Google Scholar] [CrossRef] [PubMed]
- Seol, D.W.; Li, J.; Seol, M.H.; Park, S.Y.; Talanian, R.V.; Billiar, T.R. Signaling events triggered by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL): Caspase-8 is required for TRAIL-induced apoptosis. Cancer Res. 2001, 61, 1138–1143. [Google Scholar] [PubMed]
- Tepper, C.G.; Seldin, M.F. Modulation of caspase-8 and FLICE-inhibitory protein expression as a potential mechanism of Epstein-Barr virus tumorigenesis in Burkitt’s lymphoma. Blood 1999, 94, 1727–1737. [Google Scholar] [PubMed]
- Olsson, A.; Diaz, T.; Aguilar-Santelises, M.; Osterborg, A.; Celsing, F.; Jondal, M.; Osorio, L.M. Sensitisation to TRAIL-induced apoptosis and modulation of FLICE-inhibitory protein in B chronic lymphocytic leukemia by actinomycin D. Leukemia 2001, 15, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Lee, E.W.; Seong, D.; Seo, J.; Kim, J.H.; Grootjans, S.; Kim, S.Y.; Vandenabeele, P.; Song, J. USP8 suppresses death receptor-mediated apoptosis by enhancing FLIPL stability. Oncogene 2017, 36, 458–470. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Rue, M.; Comella, J.X.; Matias-Guiu, X. FLIP is frequently expressed in endometrial carcinoma and has a role in resistance to TRAIL-induced apoptosis. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Yin, B.; Chen, J.; Ma, B.; Cai, D.; He, X. Over-expression of c-FLIP confers the resistance to TRAIL-induced apoptosis on gallbladder carcinoma. Tohoku J. Exp. Med. 2009, 217, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Grunert, M.; Gottschalk, K.; Kapahnke, J.; Gündisch, S.; Kieser, A.; Jeremias, I. The adaptor protein FADD and the initiator caspase-8 mediate activation of NF-κB by TRAIL. Cell Death Dis. 2012, 3, e414. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. CIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef]
- Keane, M.M.; Rubinstein, Y.; Cuello, M.; Ettenberg, S.A.; Banerjee, P.; Nau, M.M.; Lipkowitz, S. Inhibition of NF-kappaB activity enhances TRAIL mediated apoptosis in breast cancer cell lines. Breast Cancer Res. Treat. 2000, 64, 211–219. [Google Scholar] [CrossRef]
- Trauzold, A.; Wermann, H.; Arlt, A.; Schutze, S.; Schafer, H.; Oestern, S.; Roder, C.; Ungefroren, H.; Lampe, E.; Heinrich, M.; et al. CD95 and TRAIL receptor-mediated activation of protein kinase C and NF-kappaB contributes to apoptosis resistance in ductal pancreatic adenocarcinoma cells. Oncogene 2001, 20, 4258–4269. [Google Scholar] [CrossRef]
- Khanbolooki, S.; Nawrocki, S.T.; Arumugam, T.; Andtbacka, R.; Pino, M.S.; Kurzrock, R.; Logsdon, C.D.; Abbruzzese, J.L.; McConkey, D.J. Nuclear factor-kappaB maintains TRAIL resistance in human pancreatic cancer cells. Mol. Cancer Ther. 2006, 5, 2251–2260. [Google Scholar] [CrossRef]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Blackwell, K.; Workman, L.M.; Chen, S.; Pope, M.R.; Janz, S.; Habelhah, H. RIP1 Cleavage in the Kinase Domain Regulates TRAIL-Induced NF-kappaB Activation and Lymphoma Survival. Mol. Cell. Biol. 2015, 35, 3324–3338. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.E.; Holmstrom, T.H.; Ahonen, M.; Kahari, V.M.; Eriksson, J.E. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J. Biol. Chem. 2001, 276, 16484–16490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Borrow, J.M.; Zhang, X.Y.; Nguyen, T.; Hersey, P. Activation of ERK1/2 protects melanoma cells from TRAIL-induced apoptosis by inhibiting Smac/DIABLO release from mitochondria. Oncogene 2003, 22, 2869–2881. [Google Scholar] [CrossRef]
- Morel, J.; Audo, R.; Hahne, M.; Combe, B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J. Biol. Chem. 2005, 280, 15709–15718. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, K.; Srinivasula, S.M.; Alnemri, E.S.; Thompson, C.B.; Korsmeyer, S.J.; Bryant, J.L.; Srivastava, R.K. Involvement of proapoptotic molecules Bax and Bak in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced mitochondrial disruption and apoptosis: Differential regulation of cytochrome c and Smac/DIABLO release. Cancer Res. 2003, 63, 1712–1721. [Google Scholar] [PubMed]
- Hinz, S.; Trauzold, A.; Boenicke, L.; Sandberg, C.; Beckmann, S.; Bayer, E.; Walczak, H.; Kalthoff, H.; Ungefroren, H. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene 2000, 19, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Meyer, E.; Debatin, K.M. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002, 21, 2283–2294. [Google Scholar] [CrossRef]
- Zhu, H.; Guo, W.; Zhang, L.; Davis, J.J.; Wu, S.; Teraishi, F.; Cao, X.; Smythe, W.R.; Fang, B. Enhancing TRAIL-induced apoptosis by Bcl-X(L) siRNA. Cancer Biol. Ther. 2005, 4, 393–397. [Google Scholar] [CrossRef]
- Vogler, M.; Walczak, H.; Stadel, D.; Haas, T.L.; Genze, F.; Jovanovic, M.; Gschwend, J.E.; Simmet, T.; Debatin, K.M.; Fulda, S. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res. 2008, 68, 7956–7965. [Google Scholar] [CrossRef]
- Fakler, M.; Loeder, S.; Vogler, M.; Schneider, K.; Jeremias, I.; Debatin, K.M.; Fulda, S. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood 2009, 113, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.S.; Kim, S.H.; Ogi, K.; Plastaras, J.P.; Ling, J.; Wang, W.; Jin, Z.; Liu, Y.Y.; Dicker, D.T.; Chiao, P.J.; et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007, 12, 66–80. [Google Scholar] [CrossRef]
- Zhang, X.D.; Zhang, X.Y.; Gray, C.P.; Nguyen, T.; Hersey, P. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of human melanoma is regulated by smac/DIABLO release from mitochondria. Cancer Res. 2001, 61, 7339–7348. [Google Scholar]
- Duckett, C.S.; Nava, V.E.; Gedrich, R.W.; Clem, R.J.; Van Dongen, J.L.; Gilfillan, M.C.; Shiels, H.; Hardwick, J.M.; Thompson, C.B. A conserved family of cellular genes related to the baculovirus iap gene and encoding apoptosis inhibitors. EMBO J. 1996, 15, 2685–2694. [Google Scholar] [CrossRef]
- Ng, C.P.; Zisman, A.; Bonavida, B. Synergy is achieved by complementation with Apo2L/TRAIL and actinomycin D in Apo2L/TRAIL-mediated apoptosis of prostate cancer cells: Role of XIAP in resistance. Prostate 2002, 53, 286–299. [Google Scholar] [CrossRef]
- Ng, C.P.; Bonavida, B. X-linked inhibitor of apoptosis (XIAP) blocks Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis of prostate cancer cells in the presence of mitochondrial activation: Sensitisation by overexpression of second mitochondria-derived activator of caspase/direct IAP-binding protein with low pl (Smac/DIABLO). Mol. Cancer Ther. 2002, 1, 1051–1058. [Google Scholar] [PubMed]
- Allensworth, J.L.; Aird, K.M.; Aldrich, A.J.; Batinic-Haberle, I.; Devi, G.R. XIAP inhibition and generation of reactive oxygen species enhances TRAIL sensitivity in inflammatory breast cancer cells. Mol. Cancer Ther. 2012, 11, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Sommer, K.W.; Schamberger, C.J.; Schmidt, G.E.; Sasgary, S.; Cerni, C. Inhibitor of apoptosis protein (IAP) survivin is upregulated by oncogenic c-H-Ras. Oncogene 2003, 22, 4266–4280. [Google Scholar] [CrossRef]
- Holcik, M.; Yeh, C.; Korneluk, R.G.; Chow, T. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene 2000, 19, 4174–4177. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Nakayama, H.; Tanaka, T.; Yoshida, R.; Yoshitake, Y.; Fukuma, D.; Kawahara, K.; Nakagawa, Y.; Ota, K.; Hiraki, A.; et al. Overexpression of cIAP2 contributes to 5-FU resistance and a poor prognosis in oral squamous cell carcinoma. Br. J. Cancer 2011, 105, 1322–1330. [Google Scholar] [CrossRef]
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Smac Mimetics to Therapeutically Target IAP Proteins in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 157–169. [Google Scholar]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Fröhlich, L.F. P53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science (N. Y.) 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Pentimalli, F. Autophagy in disease: Hunger for translation. Cell Death Dis. 2019, 10, 247. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Mills, K.R.; Reginato, M.; Debnath, J.; Queenan, B.; Brugge, J.S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 3438–3443. [Google Scholar] [CrossRef]
- Das, S.; Nayak, A.; Siddharth, S.; Nayak, D.; Narayan, S.; Kundu, C.N. TRAIL enhances quinacrine-mediated apoptosis in breast cancer cells through induction of autophagy via modulation of p21 and DR5 interactions. Cell. Oncol. 2017, 40, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.I.; Ryan, K.M. Autophagy Determines the Path on the TRAIL to Death. Dev. Cell 2016, 37, 291–293. [Google Scholar] [CrossRef]
- He, W.; Wang, Q.; Xu, J.; Xu, X.; Padilla, M.T.; Ren, G.; Gou, X.; Lin, Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy 2012, 8, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Martin, G.; Hoyer-Hansen, M.; Garcia-Garcia, C.; Fumarola, C.; Farkas, T.; Lopez-Rivas, A.; Jaattela, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. A Complex between Atg7 and Caspase-9: A Novel Mechanism of Cross-regulation between Autophagy and Apoptosis. J. Biol. Chem. 2014, 289, 6485–6497. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Thorburn, J.; Andrysik, Z.; Staskiewicz, L.; Gump, J.; Maycotte, P.; Oberst, A.; Green, D.R.; Espinosa, J.M.; Thorburn, A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014, 7, 45–52. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Knoll, G.; Bittner, S.; Kurz, M.; Jantsch, J.; Ehrenschwender, M. Hypoxia regulates TRAIL sensitivity of colorectal cancer cells through mitochondrial autophagy. Oncotarget 2016, 7, 41488–41504. [Google Scholar] [CrossRef]
- Di, X.; Zhang, G.; Zhang, Y.; Takeda, K.; Rivera Rosado, L.A.; Zhang, B. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget 2013, 4, 1349–1364. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model. Cancers 2019, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Roux, J.; Hafner, M.; Bandara, S.; Sims, J.J.; Hudson, H.; Chai, D.; Sorger, P.K. Fractional killing arises from cell-to-cell variability in overcoming a caspase activity threshold. Mol. Syst. Biol. 2015, 11, 803. [Google Scholar] [CrossRef]
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459, 428–432. [Google Scholar] [CrossRef]
- Tang, W.; Wang, W.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. TRAIL receptor mediates inflammatory cytokine release in an NF-kappaB-dependent manner. Cell Res. 2009, 19, 758–767. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; O’Connell, M.P.; Ultsch, M.H.; Hurst, A.; Totpal, K.; Ashkenazi, A.; de Vos, A.M.; Kelley, R.F. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 2000, 39, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Meier, P.; Tschopp, J.; Schneider, P. Cysteine 230 is essential for the structure and activity of the cytotoxic ligand TRAIL. J. Biol. Chem. 2000, 275, 20632–20637. [Google Scholar] [CrossRef]
- Lawrence, D.; Shahrokh, Z.; Marsters, S.; Achilles, K.; Shih, D.; Mounho, B.; Hillan, K.; Totpal, K.; DeForge, L.; Schow, P.; et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat. Med. 2001, 7, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Kim, T.H.; Seol, D.W.; Esplen, J.E.; Dorko, K.; Billiar, T.R.; Strom, S.C. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 2000, 6, 564–567. [Google Scholar] [CrossRef]
- Nagata, S. Steering anti-cancer drugs away from the TRAIL. Nat. Med. 2000, 6, 502–503. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Abd El Hay, M.; Conti, A.; Arce, F.; Montinaro, A.; Papenfuss, K.; El-Bahrawy, M.A.; Walczak, H. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ. 2014, 21, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Haynes, N.M.; Hawkins, E.D.; Li, M.; McLaughlin, N.M.; Hammerling, G.J.; Schwendener, R.; Winoto, A.; Wensky, A.; Yagita, H.; Takeda, K.; et al. CD11c+ dendritic cells and B cells contribute to the tumoricidal activity of anti-DR5 antibody therapy in established tumors. J. Immunol. 2010, 185, 532–541. [Google Scholar] [CrossRef]
- Holland, P.M. Death receptor agonist therapies for cancer, which is the right TRAIL? Cytokine Growth Factor Rev. 2014, 25, 185–193. [Google Scholar] [CrossRef]
- Tuthill, M.H.; Montinaro, A.; Zinngrebe, J.; Prieske, K.; Draber, P.; Prieske, S.; Newsom-Davis, T.; von Karstedt, S.; Graves, J.; Walczak, H. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene 2015, 34, 2138–2144. [Google Scholar] [CrossRef]
- Legler, K.; Hauser, C.; Egberts, J.-H.; Willms, A.; Heneweer, C.; Boretius, S.; Röcken, C.; Glüer, C.-C.; Becker, T.; Kluge, M.; et al. The novel TRAIL-receptor agonist APG350 exerts superior therapeutic activity in pancreatic cancer cells. Cell Death Dis. 2018, 9, 445. [Google Scholar] [CrossRef]
- Hari, Y.; Harashima, N.; Tajima, Y.; Harada, M. Bcl-xL inhibition by molecular-targeting drugs sensitizes human pancreatic cancer cells to TRAIL. Oncotarget 2015, 6, 41902–41915. [Google Scholar] [CrossRef] [PubMed]
- Newsom-Davis, T.; Prieske, S.; Walczak, H. Is TRAIL the holy grail of cancer therapy? Apoptosis Int. J. Program. Cell Death 2009, 14, 607–623. [Google Scholar] [CrossRef]
- Niemoeller, O.M.; Belka, C. Radiotherapy and TRAIL for cancer therapy. Cancer Lett. 2013, 332, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H. Bortezomib. Recent results in cancer research Fortschritte der Krebsforschung Progres dans les recherches sur le cancer. Am. J. Dis. Child. 2014, 201, 325–345. [Google Scholar]
- Bychkov, M.L.; Gasparian, M.E.; Dolgikh, D.A.; Kirpichnikov, M.P. Combination of TRAIL with bortezomib shifted apoptotic signaling from DR4 to DR5 death receptor by selective internalization and degradation of DR4. PLoS ONE 2014, 9, e109756. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, X.; Qiao, J.; Bao, A. Autophagy is a regulator of TRAIL-induced apoptosis in NSCLC A549 cells. J. Cell Commun. Signal. 2017, 11, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Chao, A.; Tsai, C.L.; Chuang, W.C.; Huang, W.P.; Chen, G.C.; Lin, C.Y.; Wang, T.H.; Wang, H.S.; Lai, C.H. Bortezomib enhances cancer cell death by blocking the autophagic flux through stimulating ERK phosphorylation. Cell Death Dis. 2014, 5, e1510. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef] [PubMed]
- Geserick, P.; Drewniok, C.; Hupe, M.; Haas, T.L.; Diessenbacher, P.; Sprick, M.R.; Schon, M.P.; Henkler, F.; Gollnick, H.; Walczak, H.; et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL- and CD95L-mediated apoptosis. Oncogene 2008, 27, 3211–3220. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Giaisi, M.; Kohler, R.; Chen, W.M.; Krammer, P.H.; Li-Weber, M. Rocaglamide breaks TRAIL-resistance in human multiple myeloma and acute T-cell leukemia in vivo in a mouse xenogtraft model. Cancer Lett. 2017, 389, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Li-Weber, M. Molecular mechanisms and anti-cancer aspects of the medicinal phytochemicals rocaglamides (=flavaglines). Int. J. Cancer 2015, 137, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Nalli, A.D.; Brown, L.E.; Thomas, C.L.; Sayers, T.J.; Porco, J.A., Jr.; Henrich, C.J. Sensitisation of renal carcinoma cells to TRAIL-induced apoptosis by rocaglamide and analogs. Sci. Rep. 2018, 8, 17519. [Google Scholar] [CrossRef]
- Luan, Z.; He, Y.; He, F.; Chen, Z. Rocaglamide overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells by attenuating the inhibition of caspase-8 through cellular FLICE-like-inhibitory protein downregulation. Mol. Med. Rep. 2015, 11, 203–211. [Google Scholar] [CrossRef]
- Fulda, S. Molecular pathways: Targeting inhibitor of apoptosis proteins in cancer--from molecular mechanism to therapeutic application. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 289–295. [Google Scholar] [CrossRef]
- Fulda, S.; Wick, W.; Weller, M.; Debatin, K.M. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 2002, 8, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Thomas, R.M.; Suzuki, H.; De Brabander, J.K.; Wang, X.; Harran, P.G. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science (N. Y.) 2004, 305, 1471–1474. [Google Scholar] [CrossRef]
- Noonan, A.M.; Bunch, K.P.; Chen, J.Q.; Herrmann, M.A.; Lee, J.M.; Kohn, E.C.; O’Sullivan, C.C.; Jordan, E.; Houston, N.; Takebe, N.; et al. Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 2016, 122, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Schilder, R.J.; Albertella, M.; Strauss, J.; Sydvander, M.; Nair, S.M.; Urakpo, K.; Norin, S.; Öhd, J. A phase 1/2 study with birinapant in combination with pembrolizumab. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS3131. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Birrer, M.J.; DiCarlo, B.A.; Gaillard, S.; Martin, L.P.; Nemunaitis, J.J.; Perez, R.P.; Schilder, R.J.; Annunziata, C.M.; Begley, C.G.; et al. A phase 1b, open-label, non-randomized multicenter study of birinapant in combination with conatumumab in subjects with relapsed epithelial ovarian cancer, primary peritoneal cancer, or fallopian tube cancer. J. Clin. Oncol. 2015, 33 (Suppl. 15), 5571. [Google Scholar] [CrossRef]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Jones, J.A.; Mato, A.R.; Wierda, W.G.; Davids, M.S.; Choi, M.; Cheson, B.D.; Furman, R.R.; Lamanna, N.; Barr, P.M.; Zhou, L.; et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: An interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018, 19, 65–75. [Google Scholar] [CrossRef]
- Tolcher, A.W.; LoRusso, P.; Arzt, J.; Busman, T.A.; Lian, G.; Rudersdorf, N.S.; Vanderwal, C.A.; Kirschbrown, W.; Holen, K.D.; Rosen, L.S. Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with erlotinib in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1025–1032. [Google Scholar] [CrossRef]
- Yang, J.; Pradhan, R.S.; Rosen, L.S.; Graham, A.M.; Holen, K.D.; Xiong, H. Effect of rifampin on the pharmacokinetics, safety and tolerability of navitoclax (ABT-263), a dual inhibitor of Bcl-2 and Bcl-XL, in patients with cancer. J. Clin. Pharm. Ther. 2014, 39, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Petrungaro, A.; Uccello, G.; Vigna, E.; Recchia, A.G.; Caruso, N.; Bossio, S.; De Stefano, L.; Palummo, A.; Storino, F.; et al. Venetoclax for the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 1307–1316. [Google Scholar] [CrossRef]
- Huang, S.; Sinicrope, F.A. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res. 2008, 68, 2944–2951. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhan, Y.; Wang, H.; Li, W. ABT-263 sensitizes TRAIL-resistant hepatocarcinoma cells by downregulating the Bcl-2 family of anti-apoptotic protein. Cancer Chemother. Pharmacol. 2012, 69, 799–805. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretz, A.-L.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. TRAILblazing Strategies for Cancer Treatment. Cancers 2019, 11, 456. https://doi.org/10.3390/cancers11040456
Kretz A-L, Trauzold A, Hillenbrand A, Knippschild U, Henne-Bruns D, von Karstedt S, Lemke J. TRAILblazing Strategies for Cancer Treatment. Cancers. 2019; 11(4):456. https://doi.org/10.3390/cancers11040456
Chicago/Turabian StyleKretz, Anna-Laura, Anna Trauzold, Andreas Hillenbrand, Uwe Knippschild, Doris Henne-Bruns, Silvia von Karstedt, and Johannes Lemke. 2019. "TRAILblazing Strategies for Cancer Treatment" Cancers 11, no. 4: 456. https://doi.org/10.3390/cancers11040456
APA StyleKretz, A.-L., Trauzold, A., Hillenbrand, A., Knippschild, U., Henne-Bruns, D., von Karstedt, S., & Lemke, J. (2019). TRAILblazing Strategies for Cancer Treatment. Cancers, 11(4), 456. https://doi.org/10.3390/cancers11040456