PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise?

 , , , ,
, , , ,  and
and
Abstract
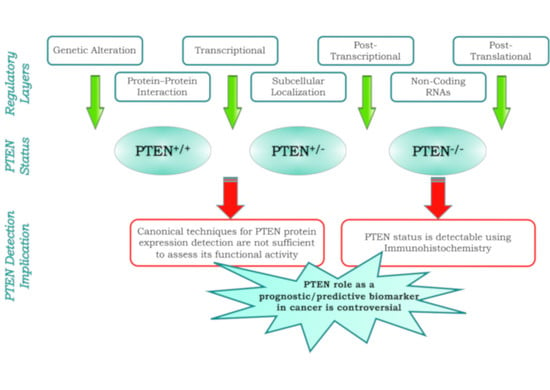
1. Introduction
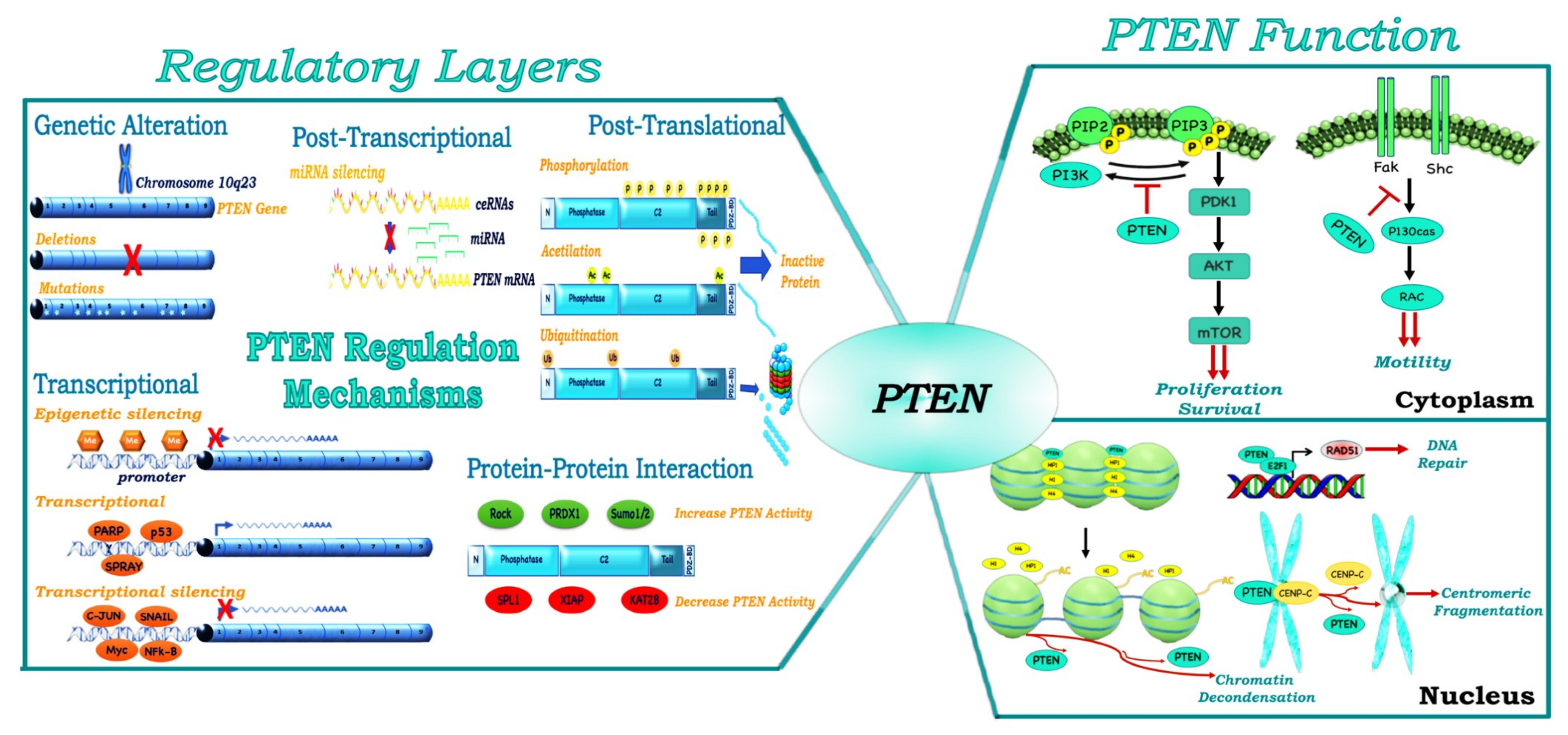
2. Multiple Layers of PTEN Regulation
3. PTEN, a Tumor Suppressor and More
4. PTEN as a Prognostic Biomarker
4.1. Urogenital Cancers
4.1.1. Prostate
4.1.2. Kidney
4.2. Gastrointestinal Cancers
4.2.1. Pancreas
4.2.2. Colorectal
4.3. Breast Cancer
4.4. Endometrial Cancer
4.5. Brain Cancers
4.6. Skin Cancers
4.7. Hematological Cancers
4.8. Pan-Cancer Overview
5. PTEN as a Predictive Biomarker
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| CCRCC | Clear cell renal cell carcinoma |
| CENP-C | Centromeric protein C |
| CIMP | CpG island methylator phenotype |
| CRC | Colorectal cancer |
| DFS | Disease-free survival |
| EC | Endometrial carcinoma |
| EEC | Endometrioid endometrial carcinoma |
| EGFR | Epidermal growth factor receptor |
| FAK | Focal adhesion kinase |
| FISH | Fluorescence in situ hybridation |
| GISTIC | Genomic identification of significant targets in cancer |
| GBM | Glioblastoma |
| IHC | Immunohistochemistry |
| IL | Interleukin |
| KIRC | Kidney renal papillary carcinoma |
| KRAS | Kirsten rat sarcoma |
| LGG | Lower grade gliomas |
| MAN2C1 | α-mannosidase 2C1 |
| MAPK | Mitogen-activated protein kinase |
| mCRC | Metastatic colorectal cancer |
| MEK | Mitogen-activated protein kinase kinase |
| miRNA | Micro RNA |
| MMP | Matrix metalloproteinase |
| MSI | Microsatellite instability |
| mTOR | Mammalian target of rapamycin |
| OS | Overall survival |
| PBD | PIP2-binding domain |
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death ligand-1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PI3K | PhosphatidylInositol 3-kinase |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
| PICT-1 | Protein interacting with carboxyl terminus 1 |
| PIP2 | PhosphatIdylinositol-4,5-bisphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| RCC | Renal cell carcinoma |
| TCGA | The cancer genome atlas |
| TMA | Tissue microarray |
| TME | Tumor microenvironment |
| TNM | Tumor, node, metastasis |
| UCEC | Uterine corpus endometrial carcinoma |
References
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The cosmic cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Kalikaki, A.; Koutsopoulos, A.; Trypaki, M.; Souglakos, J.; Stathopoulos, E.; Georgoulias, V.; Mavroudis, D.; Voutsina, A. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br. J. Cancer 2008, 99, 923–929. [Google Scholar] [CrossRef]
- Ellsworth, R.E.; Blackburn, H.L.; Shriver, C.D.; Soon-Shiong, P.; Ellsworth, D.L. Molecular heterogeneity in breast cancer: State of the science and implications for patient care. Semin. Cell Dev. Biol. 2017, 64, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Oldenhuis, C.N.; Oosting, S.F.; Gietema, J.A.; de Vries, E.G. Prognostic versus predictive value of biomarkers in oncology. Eur. J. Cancer 2008, 44, 946–953. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple functions in human malignant tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Ngeow, J.; Sesock, K.; Eng, C. Clinical implications for germline PTEN spectrum disorders. Endocrinol. Metab. Clin. North Am. 2017, 46, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Ngeow, J.; Eng, C. PTEN-opathies: From biological insights to evidence-based precision medicine. J. Clin. Investig. 2019, 129, 452–464. [Google Scholar] [CrossRef]
- Zhou, J.; Parada, L.F. PTEN signaling in autism spectrum disorders. Curr. Opin. Neurobiol. 2012, 22, 873–879. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBIO cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG s1115 study randomized clinical trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined mek and mtor inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; Del Curatolo, A.; Falcone, I.; Conciatori, F.; Bazzichetto, C.; Cognetti, F.; Corbo, V.; Scarpa, A.; Milella, M. Lack of growth inhibitory synergism with combined MAPK/PI3K inhibition in preclinical models of pancreatic cancer. Ann. Oncol. 2017, 28, 2896–2898. [Google Scholar] [CrossRef]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, D. Pi (3) king apart pten’s role in cancer. Clin. Cancer Res. 2010, 16, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in PTEN dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Correia, N.C.; Girio, A.; Antunes, I.; Martins, L.R.; Barata, J.T. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur. J. Cancer 2014, 50, 216–225. [Google Scholar] [CrossRef]
- Bermudez Brito, M.; Goulielmaki, E.; Papakonstanti, E.A. Focus on PTEN regulation. Front. Oncol. 2015, 5, 166. [Google Scholar] [CrossRef]
- Tamguney, T.; Stokoe, D. New insights into PTEN. J. Cell Sci. 2007, 120, 4071–4079. [Google Scholar] [CrossRef]
- He, L. Posttranscriptional regulation of PTEN dosage by noncoding RNAs. Sci. Signal. 2010, 3, pe39. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; Di Sanza, C.; Cesta Incani, U.; Eramo, A.; Desideri, M.; Biagioni, F.; Passeri, D.; Falcone, I.; Sette, G.; Bergamo, P.; et al. The mitogen-activated protein kinase (MAPK) cascade controls phosphatase and tensin homolog (PTEN) expression through multiple mechanisms. J. Mol. Med. (Berlin) 2012, 90, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.O.; Corcoran, M.; Grander, D.; Morris, K.V. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 2013, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Leslie, N.R.; Downes, C.P. PTEN function: How normal cells control it and tumour cells lose it. Biochem. J. 2004, 382, 1–11. [Google Scholar] [CrossRef]
- Rahdar, M.; Inoue, T.; Meyer, T.; Zhang, J.; Vazquez, F.; Devreotes, P.N. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. USA 2009, 106, 480–485. [Google Scholar] [CrossRef]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef]
- Wu, X.; Hepner, K.; Castelino-Prabhu, S.; Do, D.; Kaye, M.B.; Yuan, X.J.; Wood, J.; Ross, C.; Sawyers, C.L.; Whang, Y.E. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc. Natl. Acad. Sci. USA 2000, 97, 4233–4238. [Google Scholar] [CrossRef]
- Huang, J.; Yan, J.; Zhang, J.; Zhu, S.; Wang, Y.; Shi, T.; Zhu, C.; Chen, C.; Liu, X.; Cheng, J.; et al. Sumo1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 2012, 3, 911. [Google Scholar] [CrossRef]
- Torres, J.; Pulido, R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 2001, 276, 993–998. [Google Scholar] [CrossRef]
- Bolduc, D.; Rahdar, M.; Tu-Sekine, B.; Sivakumaren, S.C.; Raben, D.; Amzel, L.M.; Devreotes, P.; Gabelli, S.B.; Cole, P. Phosphorylation-mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife 2013, 2, e00691. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Dempsey, D.R.; Thomas, S.N.; Hayward, D.; Bolduc, D.M.; Cole, P.A. Molecular features of phosphatase and tensin homolog (PTEN) regulation by C-terminal phosphorylation. J. Biol. Chem. 2016, 291, 14160–14169. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, Z.; Zhou, S.F.; Lu, N. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des. Devel. Ther. 2014, 8, 1745–1751. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, H.Y.; Lee, J.H.; Lee, H.; Kang, G.; Song, J.S.; Kang, J.; Kim, J.H. Prognostic significance of biallelic loss of PTEN in clear cell renal cell carcinoma. J. Urol. 2014, 192, 940–946. [Google Scholar] [CrossRef]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Yim, E.K.; Peng, G.; Dai, H.; Hu, R.; Li, K.; Lu, Y.; Mills, G.B.; Meric-Bernstam, F.; Hennessy, B.T.; Craven, R.J.; et al. RAK functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell 2009, 15, 304–314. [Google Scholar] [CrossRef]
- He, L.; Fan, C.; Kapoor, A.; Ingram, A.J.; Rybak, A.P.; Austin, R.C.; Dickhout, J.; Cutz, J.C.; Scholey, J.; Tang, D. Alpha-mannosidase 2C1 attenuates PTEN function in prostate cancer cells. Nat. Commun. 2011, 2, 307. [Google Scholar] [CrossRef]
- He, L.; Ingram, A.; Rybak, A.P.; Tang, D. Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J. Clin. Investig. 2010, 120, 2094–2108. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Sotelo, N.S.; Schepens, J.T.; Valiente, M.; Hendriks, W.J.; Pulido, R. PTEN-PDZ domain interactions: Binding of PTEN to PDZ domains of PTPN13. Methods 2015, 77–78, 147–156. [Google Scholar] [CrossRef]
- Gericke, A.; Munson, M.; Ross, A.H. Regulation of the PTEN phosphatase. Gene 2006, 374, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Pass, I.; Batty, I.H.; Van der Kaay, J.; Stolarov, J.P.; Hemmings, B.A.; Wigler, M.H.; Downes, C.P.; Tonks, N.K. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA 1998, 95, 13513–13518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C.; Piccini, A.; Myers, M.P.; Van Aelst, L.; Tonks, N.K. Functional analysis of the protein phosphatase activity of PTEN. Biochem. J. 2012, 444, 457–464. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Tran, H.; Yamada, K.M. PTEN gene and integrin signaling in cancer. J. Natl. Cancer Inst. 1999, 91, 1820–1828. [Google Scholar] [CrossRef]
- Salmena, L.; Carracedo, A.; Pandolfi, P.P. Tenets of PTEN tumor suppression. Cell 2008, 133, 403–414. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef]
- Chang, C.J.; Mulholland, D.J.; Valamehr, B.; Mosessian, S.; Sellers, W.R.; Wu, H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol. Cell. Biol. 2008, 28, 3281–3289. [Google Scholar] [CrossRef]
- Shen, S.M.; Ji, Y.; Zhang, C.; Dong, S.S.; Yang, S.; Xiong, Z.; Ge, M.K.; Yu, Y.; Xia, L.; Guo, M.; et al. Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role. Nat. Commun. 2018, 9, 2392. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Uversky, V.N.; Dave, V. PTEN proteoforms in biology and disease. Cell. Mol. Life Sci. 2017, 74, 2783–2794. [Google Scholar] [CrossRef]
- Liang, H.; Chen, X.; Yin, Q.; Ruan, D.; Zhao, X.; Zhang, C.; McNutt, M.A.; Yin, Y. PTENβ is an alternatively translated isoform of PTEN that regulates rDNA transcription. Nat. Commun. 2017, 8, 14771. [Google Scholar] [CrossRef] [PubMed]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. PTEN in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.J.; Coulter, J.; Walker, S.M.; McKechnie, M.; Neisen, J.; McCabe, N.; Kennedy, R.D.; Salto-Tellez, M.; Albanese, C.; Waugh, D.J. Potentiation of inflammatory CXCL8 signalling sustains cell survival in PTEN-deficient prostate carcinoma. Eur. Urol. 2013, 64, 177–188. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Konopka, G.; Lim, K.L.; Nutt, C.L.; Bromberg, J.F.; Frank, D.A.; Mischel, P.S.; Louis, D.N.; Bonni, A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J. Neurosci. 2008, 28, 5870–5878. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in PTEN-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Xiong, Y.X.; Wu, X.T.; Shi, D.; Fan, W.; Zhou, T.; Li, Y.C.; Huang, X. Inactivation of PTEN is associated with increased angiogenesis and VEGF overexpression in gastric cancer. World J. Gastroenterol. 2004, 10, 3225–3229. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Z.; Yang, X.; Deng, K.; Cao, Y.; Chen, H.; Pan, L. Effect of wild type PTEN gene on proliferation and invasion of multiple myeloma. Int. J. Hematol. 2010, 92, 83–94. [Google Scholar] [CrossRef]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR signaling in tumor microenvironment: An overview. Int. J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN loss increases PD-l1 protein expression and affects the correlation between PD-l1 expression and clinical parameters in colorectal cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-l1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, C.; Liu, F.; Zhao, Y.; Liu, J.; Hua, Y.; Liu, J.; Huang, J.; Ge, C. A blockade of PD-l1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2017, 10, 2115–2126. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to t cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Chan, T.A. Immunotherapy and oncogenic pathways: The PTEN connection. Cancer Discov. 2016, 6, 128–129. [Google Scholar] [CrossRef]
- Golan, T.; Milella, M.; Ackerstein, A.; Berger, R. The changing face of clinical trials in the personalized medicine and immuno-oncology era: Report from the international congress on clinical trials in oncology & hemato-oncology (icto 2017). J. Exp. Clin. Cancer Res. 2017, 36, 192. [Google Scholar] [PubMed]
- Love, D.; Stratton, E.; Stocum, M. Best practices for companion diagnostic and therapeutic development: Translating between the stakeholders. N. Biotechnol. 2012, 29, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Dracopoli, N.C.; Boguski, M.S. The evolution of oncology companion diagnostics from signal transduction to immuno-oncology. Trends Pharmacol. Sci. 2017, 38, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Milne, C.P.; Cohen, J.P.; Chakravarthy, R. Market watch: Where is personalized medicine in industry heading? Nat. Rev. Drug Discov. 2015, 14, 812–813. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, II; Gelovani, J.G.; Jacoby, J.J.; Davis, S.E.; Herbst, R.S. Methodological and practical challenges for personalized cancer therapies. Nat. Rev. Clin. Oncol. 2011, 8, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dai, B. PTEN genomic deletion defines favorable prognostic biomarkers in localized prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 5430–5437. [Google Scholar]
- Mithal, P.; Allott, E.; Gerber, L.; Reid, J.; Welbourn, W.; Tikishvili, E.; Park, J.; Younus, A.; Sangale, Z.; Lanchbury, J.S.; et al. PTEN loss in biopsy tissue predicts poor clinical outcomes in prostate cancer. Int. J. Urol. 2014, 21, 1209–1214. [Google Scholar] [CrossRef]
- Chaux, A.; Peskoe, S.B.; Gonzalez-Roibon, N.; Schultz, L.; Albadine, R.; Hicks, J.; De Marzo, A.M.; Platz, E.A.; Netto, G.J. Loss of PTEN expression is associated with increased risk of recurrence after prostatectomy for clinically localized prostate cancer. Mod. Pathol. 2012, 25, 1543–1549. [Google Scholar] [CrossRef]
- Vidotto, T.; Tiezzi, D.G.; Squire, J.A. Distinct subtypes of genomic PTEN deletion size influence the landscape of aneuploidy and outcome in prostate cancer. Mol. Cytogenet. 2018, 11, 1. [Google Scholar] [CrossRef]
- Que, W.C.; Qiu, H.Q.; Cheng, Y.; Liu, M.B.; Wu, C.Y. PTEN in kidney cancer: A review and meta-analysis. Clin. Chim. Acta 2018, 480, 92–98. [Google Scholar] [CrossRef]
- Tang, L.; Li, X.; Gao, Y.; Chen, L.; Gu, L.; Chen, J.; Lyu, X.; Zhang, Y.; Zhang, X. Phosphatase and tensin homolog (PTEN) expression on oncologic outcome in renal cell carcinoma: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0179437. [Google Scholar] [CrossRef]
- Abou Youssif, T.; Fahmy, M.A.; Koumakpayi, I.H.; Ayala, F.; Al Marzooqi, S.; Chen, G.; Tamboli, P.; Squire, J.; Tanguay, S.; Sircar, K. The mammalian target of rapamycin pathway is widely activated without PTEN deletion in renal cell carcinoma metastases. Cancer 2011, 117, 290–300. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 2018, 173, 595–610. [Google Scholar] [CrossRef]
- Okami, K.; Wu, L.; Riggins, G.; Cairns, P.; Goggins, M.; Evron, E.; Halachmi, N.; Ahrendt, S.A.; Reed, A.L.; Hilgers, W.; et al. Analysis of PTEN/MMAC1 alterations in aerodigestive tract tumors. Cancer Res. 1998, 58, 509–511. [Google Scholar]
- Hill, R.; Calvopina, J.H.; Kim, C.; Wang, Y.; Dawson, D.W.; Donahue, T.R.; Dry, S.; Wu, H. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 2010, 70, 7114–7124. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Hu, R.; Yang, H.; Liu, J.; Sui, J.; Xiang, X.; Wang, F.; Chu, L.; Song, S. PTEN gene mutations correlate to poor prognosis in glioma patients: A meta-analysis. Onco Targets Ther. 2016, 9, 3485–3492. [Google Scholar]
- Feng, C.; Yao, R.; Huang, F.; Liu, X.; Nie, W. High level of PTEN expression is associated with low-grade liver metastasis and satisfactory patient survival in pancreatic cancer. Arch. Med. Res. 2011, 42, 584–588. [Google Scholar] [CrossRef]
- Foo, W.C.; Rashid, A.; Wang, H.; Katz, M.H.; Lee, J.E.; Pisters, P.W.; Wolff, R.A.; Abbruzzese, J.L.; Fleming, J.B.; Wang, H. Loss of phosphatase and tensin homolog expression is associated with recurrence and poor prognosis in patients with pancreatic ductal adenocarcinoma. Hum. Pathol. 2013, 44, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.A.; Schwock, J.; Iakovlev, V.; Pond, G.; Hedley, D.W.; Tsao, M.S. Immunohistochemical analysis of changes in signaling pathway activation downstream of growth factor receptors in pancreatic duct cell carcinogenesis. BMC Cancer 2008, 8, 43. [Google Scholar] [CrossRef]
- Siena, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Balfour, J.; Bardelli, A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 1308–1324. [Google Scholar] [CrossRef]
- Ogino, S.; Goel, A. Molecular classification and correlates in colorectal cancer. J. Mol. Diagn. 2008, 10, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Eklof, V.; Wikberg, M.L.; Edin, S.; Dahlin, A.M.; Jonsson, B.A.; Oberg, A.; Rutegard, J.; Palmqvist, R. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br. J. Cancer 2013, 108, 2153–2163. [Google Scholar] [CrossRef]
- Sawai, H.; Yasuda, A.; Ochi, N.; Ma, J.; Matsuo, Y.; Wakasugi, T.; Takahashi, H.; Funahashi, H.; Sato, M.; Takeyama, H. Loss of PTEN expression is associated with colorectal cancer liver metastasis and poor patient survival. BMC Gastroenterol. 2008, 8, 56. [Google Scholar] [CrossRef]
- Jang, K.S.; Song, Y.S.; Jang, S.H.; Min, K.W.; Na, W.; Jang, S.M.; Jun, Y.J.; Lee, K.H.; Choi, D.; Paik, S.S. Clinicopathological significance of nuclear PTEN expression in colorectal adenocarcinoma. Histopathology 2010, 56, 229–239. [Google Scholar] [CrossRef]
- Yazdani, Y.; Farazmandfar, T.; Azadeh, H.; Zekavatian, Z. The prognostic effect of PTEN expression status in colorectal cancer development and evaluation of factors affecting it: Mir-21 and promoter methylation. J. Biomed. Sci. 2016, 23, 9. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Liang, F.; Jia, Z.L.; Song, S.T.; Jiang, Z.F. PTEN mutation, methylation and expression in breast cancer patients. Oncol. Lett. 2013, 6, 161–168. [Google Scholar] [CrossRef]
- Veeraraghavan, J.; De Angelis, C.; Reis-Filho, J.S.; Pascual, T.; Prat, A.; Rimawi, M.F.; Osborne, C.K.; Schiff, R. De-escalation of treatment in her2-positive breast cancer: Determinants of response and mechanisms of resistance. Breast 2017, 34 (Suppl. 1), S19–S26. [Google Scholar] [CrossRef]
- Saal, L.H.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.B.; Maurer, M.; Koujak, S.; Ferrando, A.A.; Malmstrom, P.; Memeo, L.; et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef]
- Golmohammadi, R.; Rakhshani, M.H.; Moslem, A.R.; Pejhan, A. Prognostic role of PTEN gene expression and length of survival of breast cancer patients in the north east of Iran. Asian Pac. J. Cancer Prev. 2016, 17, 305–309. [Google Scholar] [CrossRef]
- Jones, N.; Bonnet, F.; Sfar, S.; Lafitte, M.; Lafon, D.; Sierankowski, G.; Brouste, V.; Banneau, G.; Tunon de Lara, C.; Debled, M.; et al. Comprehensive analysis of PTEN status in breast carcinomas. Int. J. Cancer 2013, 133, 323–334. [Google Scholar] [CrossRef]
- Gbelcova, H.; Bakes, P.; Priscakova, P.; Sisovsky, V.; Hojsikova, I.; Straka, L.; Konecny, M.; Markus, J.; D'Acunto, C.W.; Ruml, T.; et al. PTEN sequence analysis in endometrial hyperplasia and endometrial carcinoma in Slovak women. Anal. Cell. Pathol. (Amsterdam) 2015, 2015, 746856. [Google Scholar]
- Mutter, G.L.; Lin, M.C.; Fitzgerald, J.T.; Kum, J.B.; Baak, J.P.; Lees, J.A.; Weng, L.P.; Eng, C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J. Natl. Cancer Inst. 2000, 92, 924–930. [Google Scholar] [CrossRef]
- Yeramian, A.; Moreno-Bueno, G.; Dolcet, X.; Catasus, L.; Abal, M.; Colas, E.; Reventos, J.; Palacios, J.; Prat, J.; Matias-Guiu, X. Endometrial carcinoma: Molecular alterations involved in tumor development and progression. Oncogene 2013, 32, 403–413. [Google Scholar] [CrossRef]
- Zhang, H.M.; Fan, T.T.; Li, W.; Li, X.X. Expressions and significances of ttf-1 and PTEN in early endometrial cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 20–26. [Google Scholar]
- Raffone, A.; Travaglino, A.; Saccone, G.; Campanino, M.R.; Mollo, A.; De Placido, G.; Insabato, L.; Zullo, F. Loss of PTEN expression as diagnostic marker of endometrial precancer: A systematic review and meta-analysis. Acta Obstet. Gynecol. Scand. 2018. [Google Scholar] [CrossRef]
- James, C.D.; Galanis, E.; Frederick, L.; Kimmel, D.W.; Cunningham, J.M.; Atherton-Skaff, P.J.; O’Fallon, J.R.; Jenkins, R.B.; Buckner, J.C.; Hunter, S.B.; et al. Tumor suppressor gene alterations in malignant gliomas: Histopathological associations and prognostic evaluation. Int. J. Oncol. 1999, 15, 547–553. [Google Scholar] [CrossRef]
- Fraser, M.M.; Bayazitov, I.T.; Zakharenko, S.S.; Baker, S.J. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 2008, 151, 476–488. [Google Scholar] [CrossRef]
- Yang, J.M.; Schiapparelli, P.; Nguyen, H.N.; Igarashi, A.; Zhang, Q.; Abbadi, S.; Amzel, L.M.; Sesaki, H.; Quinones-Hinojosa, A.; Iijima, M. Characterization of PTEN mutations in brain cancer reveals that PTEN mono-ubiquitination promotes protein stability and nuclear localization. Oncogene 2017, 36, 3673–3685. [Google Scholar] [CrossRef]
- Yang, Y.; Shao, N.; Luo, G.; Li, L.; Zheng, L.; Nilsson-Ehle, P.; Xu, N. Mutations of PTEN gene in gliomas correlate to tumor differentiation and short-term survival rate. Anticancer Res. 2010, 30, 981–985. [Google Scholar]
- Sasaki, H.; Zlatescu, M.C.; Betensky, R.A.; Ino, Y.; Cairncross, J.G.; Louis, D.N. PTEN is a target of chromosome 10q loss in anaplastic oligodendrogliomas and PTEN alterations are associated with poor prognosis. Am. J. Pathol. 2001, 159, 359–367. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Bucheit, A.D.; Chen, G.; Siroy, A.; Tetzlaff, M.; Broaddus, R.; Milton, D.; Fox, P.; Bassett, R.; Hwu, P.; Gershenwald, J.E.; et al. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin. Cancer Res. 2014, 20, 5527–5536. [Google Scholar] [CrossRef]
- Giles, K.M.; Rosenbaum, B.E.; Berger, M.; Izsak, A.; Li, Y.; Illa Bochaca, I.; Vega-Saenz de Miera, E.; Wang, J.; Darvishian, F.; Zhong, H.; et al. Revisiting the clinical and biologic relevance of partial PTEN loss in melanoma. J. Investig. Dermatol. 2019, 139, 430–438. [Google Scholar] [CrossRef]
- Lahtz, C.; Stranzenbach, R.; Fiedler, E.; Helmbold, P.; Dammann, R.H. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J. Investig. Dermatol. 2010, 130, 620–622. [Google Scholar] [CrossRef]
- Morotti, A.; Panuzzo, C.; Crivellaro, S.; Carra, G.; Torti, D.; Guerrasio, A.; Saglio, G. The role of PTEN in myeloid malignancies. Hematol. Rep. 2015, 7, 5844. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.W.; Eom, J.I.; Maeng, H.Y.; Lee, S.T.; Hahn, J.S.; Ko, Y.W.; Min, Y.H. Phosphatase and tensin homologue phosphorylation in the c-terminal regulatory domain is frequently observed in acute myeloid leukaemia and associated with poor clinical outcome. Br. J. Haematol. 2003, 122, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Paganin, M.; Grillo, M.F.; Silvestri, D.; Scapinello, G.; Buldini, B.; Cazzaniga, G.; Biondi, A.; Valsecchi, M.G.; Conter, V.; Te Kronnie, G.; et al. The presence of mutated and deleted PTEN is associated with an increased risk of relapse in childhood T cell acute lymphoblastic leukaemia treated with AIEOP-BFM ALL protocols. Br. J. Haematol. 2018, 182, 705–711. [Google Scholar] [CrossRef]
- Tesio, M.; Trinquand, A.; Ballerini, P.; Hypolite, G.; Lhermitte, L.; Petit, A.; Ifrah, N.; Baruchel, A.; Dombret, H.; Macintyre, E.; et al. Age-related clinical and biological features of PTEN abnormalities in T-cell acute lymphoblastic leukaemia. Leukemia 2017, 31, 2594–2600. [Google Scholar] [CrossRef]
- Github—cBioPortal/cgdsr. Available online: https://github.com/cBioPortal/cgdsr (accessed on 19 February 2018).
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. Gistic2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Mehta, S.; Shelling, A.; Muthukaruppan, A.; Lasham, A.; Blenkiron, C.; Laking, G.; Print, C. Predictive and prognostic molecular markers for cancer medicine. Ther. Adv. Med. Oncol. 2010, 2, 125–148. [Google Scholar] [CrossRef]
- De Bono, J.S.; De Giorgi, U.; Massard, C.; Bracarda, S.; Nava Rodrigues, D.; Kocak, I.; Font, A.; Arranz Arija, J.; Shih, K.; Radavoi, G.D.; et al. PTEN loss as a predictive biomarker for the Akt inhibitor ipatasertib combined with abiraterone acetate in patients with metastatic castration-resistant prostate cancer (mCRPC). In Proceedings of the ESMO congress, European Society for Medical Oncology, Copenhagen, Denmark, 7–11 October 2016. [Google Scholar]
- Punnoose, E.A.; Ferraldeschi, R.; Szafer-Glusman, E.; Tucker, E.K.; Mohan, S.; Flohr, P.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rodrigues, D.N.; et al. PTEN loss in circulating tumour cells correlates with PTEN loss in fresh tumour tissue from castration-resistant prostate cancer patients. Br. J. Cancer 2015, 113, 1225–1233. [Google Scholar] [CrossRef]
- Fontugne, J.; Lee, D.; Cantaloni, C.; Barbieri, C.E.; Caffo, O.; Hanspeter, E.; Mazzoleni, G.; Dalla Palma, P.; Rubin, M.A.; Fellin, G.; et al. Recurrent prostate cancer genomic alterations predict response to brachytherapy treatment. Cancer Epidemiol. Biomark. Prev. 2014, 23, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef]
- Yi, Z.; Ma, F. Biomarkers of everolimus sensitivity in hormone receptor-positive breast cancer. J. Breast Cancer 2017, 20, 321–326. [Google Scholar] [CrossRef]
- Treilleux, I.; Arnedos, M.; Cropet, C.; Wang, Q.; Ferrero, J.M.; Abadie-Lacourtoisie, S.; Levy, C.; Legouffe, E.; Lortholary, A.; Pujade-Lauraine, E.; et al. Translational studies within the TAMRAD randomized GINECO trial: Evidence for mtorc1 activation marker as a predictive factor for everolimus efficacy in advanced breast cancer. Ann. Oncol. 2015, 26, 120–125. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Chen, D.; Piccart, M.; Rugo, H.S.; Burris, H.A., 3rd; Pritchard, K.I.; Campone, M.; Noguchi, S.; Perez, A.T.; Deleu, I.; et al. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: Results from bolero-2. J. Clin. Oncol. 2016, 34, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Dave, B.; Migliaccio, I.; Gutierrez, M.C.; Wu, M.F.; Chamness, G.C.; Wong, H.; Narasanna, A.; Chakrabarty, A.; Hilsenbeck, S.G.; Huang, J.; et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J. Clin. Oncol. 2011, 29, 166–173. [Google Scholar] [CrossRef]
- Fabi, A.; Metro, G.; Di Benedetto, A.; Nistico, C.; Vici, P.; Melucci, E.; Antoniani, B.; Perracchio, L.; Sperduti, I.; Milella, M.; et al. Clinical significance of PTEN and p-Akt co-expression in HER2-positive metastatic breast cancer patients treated with trastuzumab-based therapies. Oncology 2010, 78, 141–149. [Google Scholar] [CrossRef]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef]
- Loupakis, F.; Pollina, L.; Stasi, I.; Ruzzo, A.; Scartozzi, M.; Santini, D.; Masi, G.; Graziano, F.; Cremolini, C.; Rulli, E.; et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 2622–2629. [Google Scholar] [CrossRef]
- Bazzichetto, C.C.; Conciatori, F.; Falcone, I.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor-stroma interactions and response to targeted agents in preclinical models of colorectal cancer (CRC). In Proceedings of the ESMO Congress, European Society for Medical Oncology, Madrid, Spain, 8–12 September 2017. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Site | Tumor Type | Range | Average | n Sample | n TCGA Studies | Comments |
|---|---|---|---|---|---|---|
| Kidney | CCRCC | 3–5% | 4% | 1548 patients/1549 samples | 3 | Nonsense, missense, FS ins, FS del, splice, IF del |
| Non-CCRCC | 2.8–11% | 5% | 772 patients/773 samples | 5 | Nonsense, missense, FS del, IF del | |
| Prostate | Adenocarcinoma | 17–21% | 20% | 1325 patients/1326 samples | 3 | Nonsense, missense, FS ins, FS del, splice, IF del, fusion |
| Pancreas | Adenocarcinoma | 1.1–1.6% | 1.4% | 369 patients/370 samples | 2 | NA |
| Colorectal | Adenocarcinoma | 4–8% | 6% | 1506 patients/1510 samples | 3 | Nonsense, missense, FS ins, FS del, splice |
| Breast | Invasive Carcinoma | 4–11% | 7% | 3824patients/3832 samples | 4 | Nonsense, missense, FS ins, FS del, splice, IF del |
| Ovary | Serous Cystadenocarcinoma | 6–7% | 7% | 1742 patients/1754 samples | 3 | Missense, FS del, splice |
| Uterus | EC | 21% | 21% | 114 | 2 | Nonsense, missense, FS ins, FS del, splice |
| CNS–Brain | Diffuse glioma | 4–15% | 10% | 2152 patients/2168 samples | 3 | Nonsense, missense, FS ins, splice, nonstart, fusion |
| GBM | 19–32% | 22% | 1967 patients/1987 samples | 4 | Nonsense, missense, FS ins, FS del, splice, IF del | |
| Skin | Melanoma | 14–16% | 15% | 913 patients/927 samples | 2 | Nonsense, missense, FS ins, FS del, splice, IF del, fusion |
| Myeloid | AML | 1% | 1% | 200 patients/200 samples | 3 | NA |
| Thyroid | Carcinoma | 1.2% | 1.2% | 1514 patients/1523 samples | 3 | Nonsense, missense, FS del, fusion |
| Description | PTEN Loss | PTEN Del | OS p PTEN Loss | OS p PTEN Del | DFS p PTEN Loss | DFS p PTEN Del |
|---|---|---|---|---|---|---|
| Brain LGG | 1% | 22% | 2.45E-14 | 6.58E-27 | 2.15E-10 | 4.30E-24 |
| CCRCC | 1% | 18% | 3.16E-11 | 2.68E-01 | 1.85E-07 | 1.44E-01 |
| Pancreatic Adenocarcinoma | 1% | 18% | 4.70E-04 | 8.23E-01 | 2.23E-06 | 5.87E-01 |
| UCEC | 4% | 15% | 6.63E-03 | 1.78E-03 | 7.52E-05 | 1.13E-03 |
| Multiform GBM | 10% | 89% | 1.67E-02 | 1.67E-05 | 7.96E-05 | 4.26E-06 |
| Sarcoma | 6% | 54% | 1.04E-02 | 1.28E-01 | 6.73E-04 | 1.25E-01 |
| Head and Neck Squamous Cell Carcinoma | 3% | 27% | 3.45E-01 | 7.66E-01 | 4.73E-02 | 7.55E-01 |
| Prostate Adenocarcinoma | 19% | 32% | 8.59E-01 | 8.33E-01 | 1.31E-01 | 7.68E-02 |
| Bladder Urothelial Carcinoma | 2% | 43% | 2.59E-01 | 9.70E-01 | 2.68E-01 | 8.37E-01 |
| Breast Invasive Carcinoma | 5% | 30% | 5.72E-01 | 4.17E-01 | 2.82E-01 | 5.97E-02 |
| Diffuse Large B-cell Lymphoma | 6% | 15% | 2.91E-01 | 3.95E-01 | 4.64E-01 | 6.38E-01 |
| Liver Hepatocellular Carcinoma | 4% | 26% | 9.83E-01 | 2.52E-01 | 4.92E-01 | 2.09E-01 |
| Thyroid Carcinoma | 1% | 3% | 7.20E-01 | 5.57E-01 | 5.02E-01 | 3.48E-01 |
| Testicular Germ Cell Cancer | 1% | 46% | 8.85E-01 | 3.19E-01 | 5.33E-01 | 2.01E-01 |
| Colorectal Adenocarcinoma | 3% | 26% | 1.93E-01 | 1.52E-01 | 5.33E-01 | 1.02E-01 |
| Lung Squamous Cell Carcinoma | 10% | 54% | 7.25E-02 | 1.79E-01 | 5.70E-01 | 2.35E-01 |
| Skin Cutaneous Melanoma | 7% | 63% | 2.11E-01 | 2.71E-01 | 6.04E-01 | 5.46E-02 |
| Papillary Thyroid Carcinoma | 1% | 2% | 6.85E-01 | 6.57E-01 | 6.08E-01 | 5.13E-01 |
| Kidney Renal Papillary Cell Carcinoma | 0% | 7% | 7.59E-01 | 9.27E-03 | 6.75E-01 | 1.28E-02 |
| Stomach Adenocarcinoma | 5% | 27% | 1.96E-02 | 4.46E-02 | 6.75E-01 | 5.17E-02 |
| Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma | 5% | 30% | 6.45E-01 | 2.33E-01 | 6.92E-01 | 2.05E-01 |
| Esophageal Carcinoma | 4% | 37% | 5.00E-01 | 9.30E-01 | 7.20E-01 | 1.88E-01 |
| Kidney Chromophobe | 2% | 74% | 1.00E+00 | 1.70E-01 | 7.39E-01 | 4.74E-01 |
| Lung Adenocarcinoma | 1% | 28% | 3.02E-01 | 3.32E-01 | 7.59E-01 | 4.51E-01 |
| Ovarian Serous Cystadenocarcinoma | 4% | 39% | 4.74E-01 | 7.10E-02 | 9.93E-01 | 4.56E-01 |
| AML | 1% | 2% | 2.83E-07 | 3.72E-10 | NA | NA |
| Adrenocortical Carcinoma | 0% | 11% | NA | 2.33E-01 | NA | 3.50E-01 |
| Cholangiocarcinoma | 0% | 17% | NA | 8.36E-01 | NA | 9.60E-01 |
| Esophagus-Stomach Cancers | 7% | 40% | 9.99E-01 | 5.45E-01 | NA | NA |
| Thymoma | 0% | 3% | NA | 6.27E-01 | NA | 6.04E-01 |
| Uveal Melanoma | 0% | 1% | NA | 6.86E-05 | NA | NA |
| Description of Patients | OS Median | DFS Median | |||||
|---|---|---|---|---|---|---|---|
| Tumor Type (TCGA, Provisional) | N | Control | PTEN Loss | PTEN Del | Control | PTEN Loss | PTEN Del |
| AML | 191 | 15.97 | 0.495 | 0 | NA | NA | NA |
| Adrenocortical Carcinoma | 90 | 15.97 | 0.495 | 60.84 | 68.96 | NA | 31.31 |
| Brain LGG | 513 | 105.12 | 23.88 | 24.9 | 72.17 | 14.13 | 15.97 |
| Multiform GMB | 577 | 17.77 | 12.42 | 13.96 | 14.42 | 6.21 | 7.13 |
| Head and Neck Squamous Cell Carcinoma | 522 | 54.89 | NA | 56.9 | 71.22 | NA | 61.07 |
| CCRCC | 528 | 90.8 | 3.61 | 84.23 | 123.72 | 2.04 | 88.21 |
| Kidney Renal Papillary Cell Carcinoma | 288 | NA | NA | NA | 106.04 | NA | 91.59 |
| Lung Squamous Cell Carcinoma | 501 | 48.78 | 88.04 | 56.27 | 57.72 | 146.88 | 62.81 |
| Pancreatic Adenocarcinoma | 184 | 20.17 | 15.11 | 19.94 | 17.28 | 3.22 | 14.45 |
| Sarcoma | 257 | 76.35 | 53.45 | 63.76 | 42.81 | 11.79 | 32.03 |
| Skin Cutaneous Melanoma | 367 | 74.67 | 268.53 | 107.06 | 44.65 | 66.03 | 56.8 |
| Stomach Adenocarcinoma | 441 | 55.39 | 18.33 | 21.98 | 55.06 | 14.09 | 31.6 |
| UCEC | 539 | 112.45 | 77.27 | 102.23 | NA | 62.98 | 69.42 |
| Uveal Melanoma | 80 | 112.45 | NA | 19.91 | NA | 26.77 | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. https://doi.org/10.3390/cancers11040435
Bazzichetto C, Conciatori F, Pallocca M, Falcone I, Fanciulli M, Cognetti F, Milella M, Ciuffreda L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers. 2019; 11(4):435. https://doi.org/10.3390/cancers11040435
Chicago/Turabian StyleBazzichetto, Chiara, Fabiana Conciatori, Matteo Pallocca, Italia Falcone, Maurizio Fanciulli, Francesco Cognetti, Michele Milella, and Ludovica Ciuffreda. 2019. "PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise?" Cancers 11, no. 4: 435. https://doi.org/10.3390/cancers11040435
APA StyleBazzichetto, C., Conciatori, F., Pallocca, M., Falcone, I., Fanciulli, M., Cognetti, F., Milella, M., & Ciuffreda, L. (2019). PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers, 11(4), 435. https://doi.org/10.3390/cancers11040435