The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis


Abstract
1. Introduction
2. Identification of PCSCs
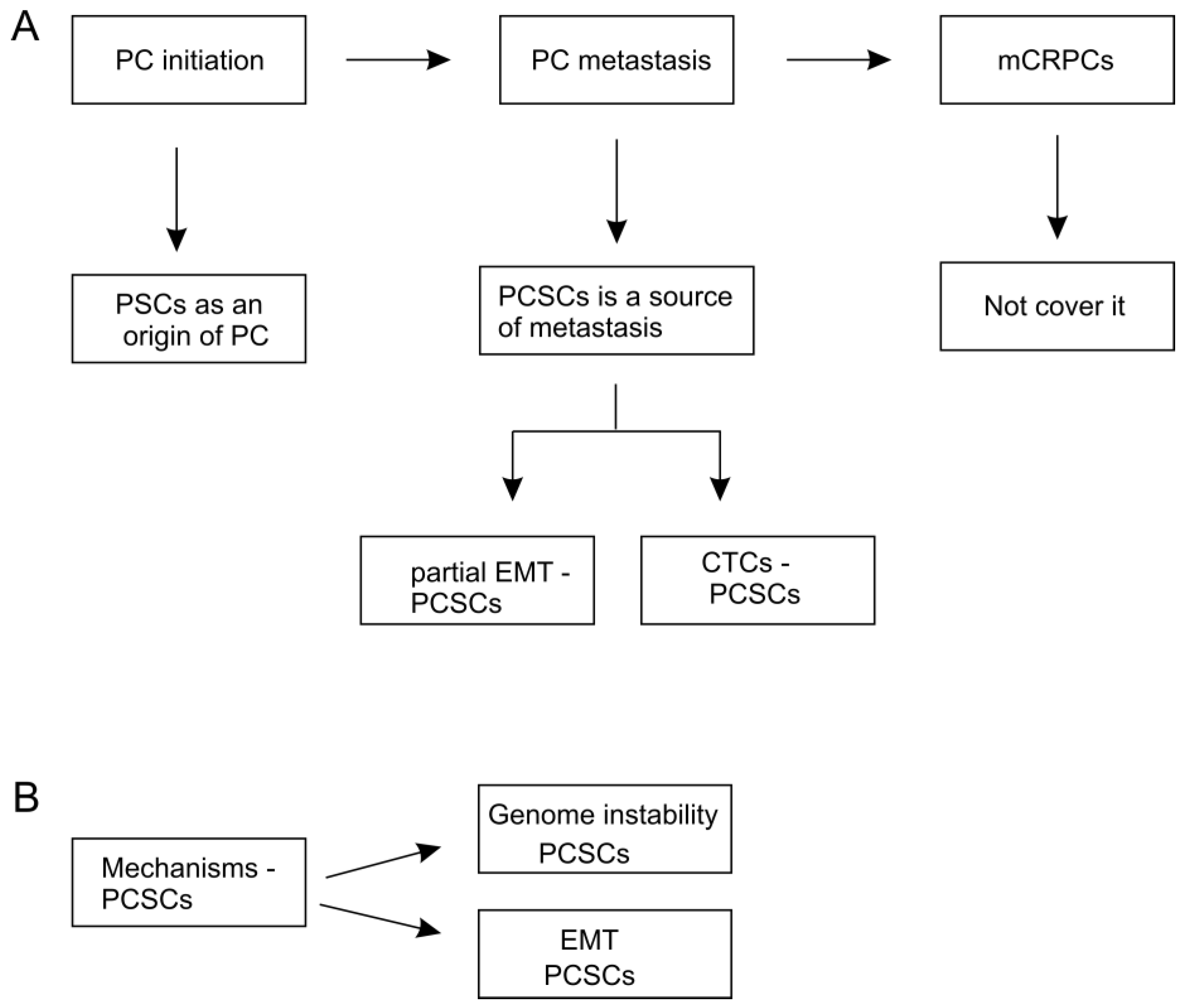
3. PCSCs as a Potential Origin of PC
4. PCSCs as a Source of PC Metastasis
4.1. The Contributions of EMT in PCSCs-Participated Metastasis
4.2. PCSCs in Circulating Tumor Cells (CTC) Lead to Metastasis
4.3. Association of PCSCs with Metastasis
5. Pathways Regulating CSCs
5.1. Dynamically Maintaining CSC “Stemness”
5.2. Mechanisms Regulating CSC Plasticity
5.3. Molecular Basis of CSC Stemness and Plasticity
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 2011, 61, 212–236. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef]
- Isaacs, J.T.; Coffey, D.S. Etiology and disease process of benign prostatic hyperplasia. Prostate. Suppl. 1989, 2, 33–50. [Google Scholar] [CrossRef]
- Lytton, B. Prostate cancer: A brief history and the discovery of hormonal ablation treatment. J. Urol. 2001, 165, 1859–1862. [Google Scholar] [CrossRef]
- Wong, Y.N.; Ferraldeschi, R.; Attard, G.; de Bono, J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol. 2014, 11, 365–376. [Google Scholar] [CrossRef]
- Skvortsov, S.; Skvortsova, I.I.; Tang, D.G.; Dubrovska, A. Concise review: Prostate cancer stem cells: Current understanding. Stem Cells 2018, 36, 1457–1474. [Google Scholar] [CrossRef]
- Ross, J.S. The androgen receptor in prostate cancer: Therapy target in search of an integrated diagnostic test. Adv. Anat. Pathol. 2007, 14, 353–357. [Google Scholar] [CrossRef]
- Moon, C.; Park, J.C.; Chae, Y.K.; Yun, J.H.; Kim, S. Current status of experimental therapeutics for prostate cancer. Cancer Lett. 2008, 266, 116–134. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, P. Diagnosis and treatment of prostate cancer. Nurs. Times 2014, 110, 12–15. [Google Scholar] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Berthold, D.R.; Pond, G.R.; Soban, F.; de Wit, R.; Eisenberger, M.; Tannock, I.F. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: Updated survival in the tax 327 study. J. Clin. Oncol. 2008, 26, 242–245. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- She, J.J.; Zhang, P.G.; Wang, Z.M.; Gan, W.M.; Che, X.M. Identification of side population cells from bladder cancer cells by dyecycle violet staining. Cancer Biol. Ther. 2008, 7, 1663–1668. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ovalle, S.; Gutierrez-Lopez, M.D.; Olmo, N.; Turnay, J.; Lizarbe, M.A.; Majano, P.; Molina-Jimenez, F.; Lopez-Cabrera, M.; Yanez-Mo, M.; Sanchez-Madrid, F.; et al. The tetraspanin CD9 inhibits the proliferation and tumorigenicity of human colon carcinoma cells. Int. J. Cancer 2007, 121, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Wu, C.; Wei, Q.; Utomo, V.; Nadesan, P.; Whetstone, H.; Kandel, R.; Wunder, J.S.; Alman, B.A. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 2007, 67, 8216–8222. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Richardson, G.D.; Robson, C.N.; Lang, S.H.; Neal, D.E.; Maitland, N.J.; Collins, A.T. Cd133, a novel marker for human prostatic epithelial stem cells. J. Cell Sci. 2004, 117, 3539–3545. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Huss, W.J.; Gray, D.R.; Greenberg, N.M.; Mohler, J.L.; Smith, G.J. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer Res. 2005, 65, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.S.; Lawson, D.A.; Cheng, D.; Sun, W.; Garraway, I.P.; Witte, O.N. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc. Natl. Acad. Sci. USA 2008, 105, 20882–20887. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Hindoyan, A.; Wang, S.; Tran, L.M.; Goldstein, A.S.; Lawson, D.; Chen, D.; Li, Y.; Guo, C.; Zhang, B.; et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS ONE 2012, 7, e42564. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Q.; Liu, X.; Liu, C.; Liu, R.; Rycaj, K.; Zhang, D.; Liu, B.; Jeter, C.; Calhoun-Davis, T.; et al. Defining a population of stem-like human prostate cancer cells that can generate and propagate castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 4505–4516. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, X.; Laffin, B.; Chen, X.; Choy, G.; Jeter, C.R.; Calhoun-Davis, T.; Li, H.; Palapattu, G.S.; Pang, S.; et al. The psa(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell 2012, 10, 556–569. [Google Scholar] [CrossRef]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Ingram, A.J.; Tang, D. Propagation of human prostate cancer stem-like cells occurs through egfr-mediated erk activation. PLoS ONE 2013, 8, e61716. [Google Scholar] [CrossRef]
- Rybak, A.P.; He, L.; Kapoor, A.; Cutz, J.C.; Tang, D. Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochim. Biophys. Acta 2011, 1813, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Tang, D. SOX2 plays a critical role in egfr-mediated self-renewal of human prostate cancer stem-like cells. Cell. Signal. 2013, 25, 2734–2742. [Google Scholar] [CrossRef]
- Garraway, I.P.; Sun, W.; Tran, C.P.; Perner, S.; Zhang, B.; Goldstein, A.S.; Hahm, S.A.; Haider, M.; Head, C.S.; Reiter, R.E.; et al. Human prostate sphere-forming cells represent a subset of basal epithelial cells capable of glandular regeneration in vivo. Prostate 2010, 70, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Rautenberg, C.; Germing, U.; Haas, R.; Kobbe, G.; Schroeder, T. Relapse of acute myeloid leukemia after allogeneic stem cell transplantation: Prevention, detection, and treatment. Int. J. Mol. Sci. 2019, 20, 228. [Google Scholar] [CrossRef]
- Wojno, K.J.; Epstein, J.I. The utility of basal cell-specific anti-cytokeratin antibody (34 beta e12) in the diagnosis of prostate cancer. A review of 228 cases. Am. J. Surg. Pathol. 1995, 19, 251–260. [Google Scholar] [CrossRef]
- Shah, R.B.; Zhou, M.; LeBlanc, M.; Snyder, M.; Rubin, M.A. Comparison of the basal cell-specific markers, 34betae12 and p63, in the diagnosis of prostate cancer. Am. J. Surg. Pathol. 2002, 26, 1161–1168. [Google Scholar] [CrossRef]
- Grisanzio, C.; Signoretti, S. P63 in prostate biology and pathology. J. Cell Biochem. 2008, 103, 1354–1368. [Google Scholar] [CrossRef]
- Humphrey, P.A. Diagnosis of adenocarcinoma in prostate needle biopsy tissue. J. Clin. Pathol. 2007, 60, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Tsubura, A.; Okamura, A.; Senzaki, H.; Naka, Y.; Komatz, Y.; Morii, S. Keratin profiles in normal/hyperplastic prostates and prostate carcinoma. Virchows Archiv. Apathol. Anat. Histopathol. 1992, 421, 157–161. [Google Scholar] [CrossRef]
- Parsons, J.K.; Gage, W.R.; Nelson, W.G.; De Marzo, A.M. P63 protein expression is rare in prostate adenocarcinoma: Implications for cancer diagnosis and carcinogenesis. Urology 2001, 58, 619–624. [Google Scholar] [CrossRef]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a cell of origin for human prostate cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Xin, L.; Lukacs, R.U.; Cheng, D.; Witte, O.N. Isolation and functional characterization of murine prostate stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.E.; Xiong, X.; Coetzee, S.; Salm, S.N.; Moscatelli, D.; Goto, K.; Wilson, E.L. Sca-1 expression identifies stem cells in the proximal region of prostatic ducts with high capacity to reconstitute prostatic tissue. Proc. Natl. Acad. Sci. USA 2005, 102, 7180–7185. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Wang, B.E.; Johnson, L.; Gao, W.Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808. [Google Scholar] [CrossRef]
- Xin, L.; Lawson, D.A.; Witte, O.N. The sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 6942–6947. [Google Scholar] [CrossRef]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Xin, L.; Morim, A.; Lawson, D.; Witte, O.; Wu, H. Lin-sca-1+cd49fhigh stem/progenitors are tumor-initiating cells in the pten-null prostate cancer model. Cancer Res. 2009, 69, 8555–8562. [Google Scholar] [CrossRef]
- Wang, S.; Garcia, A.J.; Wu, M.; Lawson, D.A.; Witte, O.N.; Wu, H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1480–1485. [Google Scholar] [CrossRef]
- Kwon, O.J.; Xin, L. Prostate epithelial stem and progenitor cells. Am. J. Clin. Exp. Urol. 2014, 2, 209–218. [Google Scholar]
- Ousset, M.; Van Keymeulen, A.; Bouvencourt, G.; Sharma, N.; Achouri, Y.; Simons, B.D.; Blanpain, C. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat. Cell Biol. 2012, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.L.; Huang, Y.F.; You, L.R.; Chao, N.C.; Su, F.Y.; Chang, J.L.; Chen, C.M. Conditionally ablated pten in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice. Am. J. Pathol. 2013, 182, 975–991. [Google Scholar] [CrossRef]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.J.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951–961. [Google Scholar] [CrossRef]
- Choi, N.; Zhang, B.; Zhang, L.; Ittmann, M.; Xin, L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 2012, 21, 253–265. [Google Scholar] [CrossRef]
- Wang, Z.A.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal cells are favored as the cell of origin for prostate cancer. Cell Rep. 2014, 8, 1339–1346. [Google Scholar] [CrossRef]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef]
- Yoo, Y.A.; Roh, M.; Naseem, A.F.; Lysy, B.; Desouki, M.M.; Unno, K.; Abdulkadir, S.A. Bmi1 marks distinct castration-resistant luminal progenitor cells competent for prostate regeneration and tumour initiation. Nat. Commun. 2016, 7, 12943. [Google Scholar] [CrossRef]
- Yoo, Y.A.; Vatapalli, R.; Lysy, B.; Mok, H.; Desouki, M.M.; Abdulkadir, S.A. The role of castration-resistant Bmi1+ Sox2+ cells in driving recurrence in prostate cancer. J. Natl. Cancer Inst. 2019, 111, 311–321. [Google Scholar] [CrossRef]
- Fan, C.; He, L.; Kapoor, A.; Gillis, A.; Rybak, A.P.; Cutz, J.C.; Tang, D. Bmi1 promotes prostate tumorigenesis via inhibiting p16(INK4A) and p14(ARF) expression. Biochim. Biophys. Acta 2008, 1782, 642–648. [Google Scholar] [CrossRef]
- Lin, X.; Ojo, D.; Wei, F.; Wong, N.; Gu, Y.; Tang, D. A novel aspect of tumorigenesis-bmi1 functions in regulating DNA damage response. Biomolecules 2015, 5, 3396–3415. [Google Scholar] [CrossRef]
- Jia, X.; Li, X.; Xu, Y.; Zhang, S.; Mou, W.; Liu, Y.; Liu, Y.; Lv, D.; Liu, C.H.; Tan, X.; et al. Sox2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. J. Mol. Cell Biol. 2011, 3, 230–238. [Google Scholar] [CrossRef]
- Sattler, H.P.; Lensch, R.; Rohde, V.; Zimmer, E.; Meese, E.; Bonkhoff, H.; Retz, M.; Zwergel, T.; Bex, A.; Stoeckle, M.; et al. Novel amplification unit at chromosome 3q25-q27 in human prostate cancer. Prostate 2000, 45, 207–215. [Google Scholar] [CrossRef]
- Barros-Silva, J.D.; Linn, D.E.; Steiner, I.; Guo, G.; Ali, A.; Pakula, H.; Ashton, G.; Peset, I.; Brown, M.; Clarke, N.W.; et al. Single-cell analysis identifies ly6d as a marker linking castration-resistant prostate luminal cells to prostate progenitors and cancer. Cell Rep. 2018, 25, 3504–3518 e3506. [Google Scholar] [CrossRef]
- Stoyanova, T.; Cooper, A.R.; Drake, J.M.; Liu, X.; Armstrong, A.J.; Pienta, K.J.; Zhang, H.; Kohn, D.B.; Huang, J.; Witte, O.N.; et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20111–20116. [Google Scholar] [CrossRef]
- Dekoninck, S.; Blanpain, C. Stem cell dynamics, migration and plasticity during wound healing. Nat. Cell Biol. 2019, 21, 18–24. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic plasticity: Driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Gupta, R.G.; Somer, R.A. Intratumor heterogeneity: Novel approaches for resolving genomic architecture and clonal evolution. Mol. Cancer Res. 2017, 15, 1127–1137. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA damage in stem cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef]
- Visvader, J.E.; Clevers, H. Tissue-specific designs of stem cell hierarchies. Nat. Cell Biol. 2016, 18, 349–355. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Weigelt, B.; Peterse, J.L.; van’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Chiang, A.C.; Massague, J. Molecular basis of metastasis. N. Engl. J. Med. 2008, 359, 2814–2823. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining emt during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (met) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Blanpain, C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. Emt, cscs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. Emt and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. Emt transition states during tumor progression and metastasis. Trends Cell Biol. 2018. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-type-specific chromatin states differentially prime squamous cell carcinoma tumor-initiating cells for epithelial to mesenchymal transition. Cell Stem Cell 2017, 20, 191–204.e5. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during emt. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Xu, L.; Mao, X.; Guo, T.; Chan, P.Y.; Shaw, G.; Hines, J.; Stankiewicz, E.; Wang, Y.; Oliver, R.T.D.; Ahmad, A.S.; et al. The novel association of circulating tumor cells and circulating megakaryocytes with prostate cancer prognosis. Clin. Cancer Res. 2017, 23, 5112–5122. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The emt-activator zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Chang, Y.S.; Chen, W.Y.; Yin, J.J.; Sheppard-Tillman, H.; Huang, J.; Liu, Y.N. Egf receptor promotes prostate cancer bone metastasis by downregulating mir-1 and activating twist1. Cancer Res. 2015, 75, 3077–3086. [Google Scholar] [CrossRef]
- Malek, R.; Gajula, R.P.; Williams, R.D.; Nghiem, B.; Simons, B.W.; Nugent, K.; Wang, H.; Taparra, K.; Lemtiri-Chlieh, G.; Yoon, A.R.; et al. Twist1-wdr5-hottip regulates hoxa9 chromatin to facilitate prostate cancer metastasis. Cancer Res. 2017, 77, 3181–3193. [Google Scholar] [CrossRef] [PubMed]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Thiyagarajan, S.; Kato, Y.; Aziz, K.; Wang, R.; Gandhi, N.; Wild, A.T.; Vesuna, F.; et al. The twist box domain is required for twist1-induced prostate cancer metastasis. Mol. Cancer Res. 2013, 11, 1387–1400. [Google Scholar] [CrossRef]
- Ezponda, T.; Popovic, R.; Shah, M.Y.; Martinez-Garcia, E.; Zheng, Y.; Min, D.J.; Will, C.; Neri, A.; Kelleher, N.L.; Yu, J.; et al. The histone methyltransferase mmset/whsc1 activates twist1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 2013, 32, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Kogan-Sakin, I.; Tabach, Y.; Buganim, Y.; Molchadsky, A.; Solomon, H.; Madar, S.; Kamer, I.; Stambolsky, P.; Shelly, A.; Goldfinger, N.; et al. Mutant p53(r175h) upregulates twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011, 18, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic roles for zeb1 in cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef]
- Selth, L.A.; Das, R.; Townley, S.L.; Coutinho, I.; Hanson, A.R.; Centenera, M.M.; Stylianou, N.; Sweeney, K.; Soekmadji, C.; Jovanovic, L.; et al. A zeb1-mir-375-yap1 pathway regulates epithelial plasticity in prostate cancer. Oncogene 2017, 36, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Celiktas, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.M.; Nelson, P.S.; et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and src family kinases. Am. J. Pathol. 2011, 179, 400–410. [Google Scholar] [CrossRef]
- Furth, J.; Kahn, M.C.; Breedis, C. The transmission of leukemia of mice with a single cell. Am. J. Cancer 1937, 31, 276–282. [Google Scholar]
- Ashworth, T. A case of cancer in which cells similar to those in tumors were seen in the blood after death. Aust. Med. J. 1869, 14, 146–149. [Google Scholar]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1989, 133, 571–573. [Google Scholar] [CrossRef]
- Vanharanta, S.; Massague, J. Origins of metastatic traits. Cancer Cell 2013, 24, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Gross, S.; O’Hara, S.M.; Smirnov, D.A.; Terstappen, L.W.; Allard, W.J.; Bilbee, M.; Cheng, J.D.; Hoffman, J.P.; et al. Isolation and characterization of circulating tumor cells in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 2006, 6, 125–132. [Google Scholar] [CrossRef]
- Krebs, M.G.; Sloane, R.; Priest, L.; Lancashire, L.; Hou, J.M.; Greystoke, A.; Ward, T.H.; Ferraldeschi, R.; Hughes, A.; Clack, G.; et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 1556–1563. [Google Scholar] [CrossRef]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef]
- Liong, M.L.; Lim, C.R.; Yang, H.; Chao, S.; Bong, C.W.; Leong, W.S.; Das, P.K.; Loh, C.S.; Lau, B.E.; Yu, C.G.; et al. Blood-based biomarkers of aggressive prostate cancer. PLoS ONE 2012, 7, e45802. [Google Scholar] [CrossRef]
- Zhou, L.; Dicker, D.T.; Matthew, E.; El-Deiry, W.S.; Alpaugh, R.K. Circulating tumor cells: Silent predictors of metastasis. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Van Schaijik, B.; Wickremesekera, A.C.; Mantamadiotis, T.; Kaye, A.H.; Tan, S.T.; Stylli, S.S.; Itinteang, T. Circulating tumor stem cells and glioblastoma: A review. J. Clin. Neurosci. 2019, 61, 5–9. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Yang, M.H.; Imrali, A.; Heeschen, C. Circulating cancer stem cells: The importance to select. Chin. J. Cancer Res. Chung-Kuo Yen Cheng Yen Chiu 2015, 27, 437–449. [Google Scholar]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer ctcs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra148. [Google Scholar] [CrossRef]
- Grillet, F.; Bayet, E.; Villeronce, O.; Zappia, L.; Lagerqvist, E.L.; Lunke, S.; Charafe-Jauffret, E.; Pham, K.; Molck, C.; Rolland, N.; et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 2017, 66, 1802–1810. [Google Scholar] [CrossRef]
- Day, K.C.; Lorenzatti Hiles, G.; Kozminsky, M.; Dawsey, S.J.; Paul, A.; Broses, L.J.; Shah, R.; Kunja, L.P.; Hall, C.; Palanisamy, N.; et al. Her2 and egfr overexpression support metastatic progression of prostate cancer to bone. Cancer Res. 2017, 77, 74–85. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef]
- Harris, K.S.; Kerr, B.A. Prostate cancer stem cell markers drive progression, therapeutic resistance, and bone metastasis. Stem Cells Int. 2017, 2017, 8629234. [Google Scholar] [CrossRef]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.; Law, W.L.; Chu, A.C.; Poon, J.T.; Lam, C.S.; Chow, A.K.; Ng, L.; Cheung, L.W.; Lan, X.R.; Lan, H.Y.; et al. A subpopulation of cd26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar] [CrossRef]
- Saur, D.; Seidler, B.; Schneider, G.; Algul, H.; Beck, R.; Senekowitsch-Schmidtke, R.; Schwaiger, M.; Schmid, R.M. Cxcr4 expression increases liver and lung metastasis in a mouse model of pancreatic cancer. Gastroenterology 2005, 129, 1237–1250. [Google Scholar] [CrossRef]
- Juarez, J.; Bendall, L. Sdf-1 and cxcr4 in normal and malignant hematopoiesis. Histol. Histopathol. 2004, 19, 299–309. [Google Scholar]
- Karpova, D.; Bonig, H. Concise review: Cxcr4/cxcl12 signaling in immature hematopoiesis--lessons from pharmacological and genetic models. Stem Cells 2015, 33, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Domanska, U.M.; Kruizinga, R.C.; Nagengast, W.B.; Timmer-Bosscha, H.; Huls, G.; de Vries, E.G.; Walenkamp, A.M. A review on cxcr4/cxcl12 axis in oncology: No place to hide. Eur. J. Cancer 2013, 49, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.; Tavano, F.; Morelli, F.; Latiano, T.P.; Di Sebastiano, P.; Maiello, E. Chemokine receptor cxcr4: Role in gastrointestinal cancer. Crit. Rev. Oncol./Hematol. 2013, 88, 696–705. [Google Scholar] [CrossRef]
- Su, L.; Zhang, J.; Xu, H.; Wang, Y.; Chu, Y.; Liu, R.; Xiong, S. Differential expression of cxcr4 is associated with the metastatic potential of human non-small cell lung cancer cells. Clin. Cancer Res. 2005, 11, 8273–8280. [Google Scholar] [CrossRef]
- Smith, M.C.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. Cxcr4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/cxcr4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Jinnah, A.H.; Zacks, B.C.; Gwam, C.U.; Kerr, B.A. Emerging and established models of bone metastasis. Cancers 2018, 10, 176. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 1795. [Google Scholar] [CrossRef] [PubMed]
- Zhau, H.E.; He, H.; Wang, C.Y.; Zayzafoon, M.; Morrissey, C.; Vessella, R.L.; Marshall, F.F.; Chung, L.W.; Wang, R. Human prostate cancer harbors the stem cell properties of bone marrow mesenchymal stem cells. Clin. Cancer Res. 2011, 17, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Miki, J.; Furusato, B.; Li, H.; Gu, Y.; Takahashi, H.; Egawa, S.; Sesterhenn, I.A.; McLeod, D.G.; Srivastava, S.; Rhim, J.S. Identification of putative stem cell markers, cd133 and cxcr4, in htert-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007, 67, 3153–3161. [Google Scholar] [CrossRef]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. Nanog promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Cackowski, F.C.; Yumoto, K.; Decker, A.M.; Wang, J.; Kim, J.K.; Lee, E.; Wang, Y.; Chung, J.S.; Gursky, A.M.; et al. Cxcl12gamma promotes metastatic castration-resistant prostate cancer by inducing cancer stem cell and neuroendocrine phenotypes. Cancer Res. 2018, 78, 2026–2039. [Google Scholar] [CrossRef]
- Kucia, M.; Reca, R.; Miekus, K.; Wanzeck, J.; Wojakowski, W.; Janowska-Wieczorek, A.; Ratajczak, J.; Ratajczak, M.Z. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: Pivotal role of the sdf-1-cxcr4 axis. Stem Cells 2005, 23, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, R.; Zhang, D.; Deng, Q.; Liu, B.; Chao, H.P.; Rycaj, K.; Takata, Y.; Lin, K.; Lu, Y.; et al. Microrna-141 suppresses prostate cancer stem cells and metastasis by targeting a cohort of pro-metastasis genes. Nat. Commun. 2017, 8, 14270. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microrna mir-34a inhibits prostate cancer stem cells and metastasis by directly repressing cd44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Ojo, D.; Lin, X.; Wong, N.; Gu, Y.; Tang, D. Prostate cancer stem-like cells contribute to the development of castration-resistant prostate cancer. Cancers 2015, 7, 2290–2308. [Google Scholar] [CrossRef]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Anti-cd38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. Cd133(+) and cd133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Kanwal, R.; Shukla, S.; Walker, E.; Gupta, S. Acquisition of tumorigenic potential and therapeutic resistance in cd133+ subpopulation of prostate cancer cells exhibiting stem-cell like characteristics. Cancer Lett. 2018, 430, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, H.; Cannon, V.; Wolcott, K.M.; Song, H.; Yates, C. Side population rather than cd133(+) cells distinguishes enriched tumorigenicity in htert-immortalized primary prostate cancer cells. Mol. Cancer 2011, 10, 112. [Google Scholar] [CrossRef]
- Moreb, J.S.; Baker, H.V.; Chang, L.J.; Amaya, M.; Lopez, M.C.; Ostmark, B.; Chou, W. Aldh isozymes downregulation affects cell growth, cell motility and gene expression in lung cancer cells. Mol. Cancer 2008, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Douville, J.; Beaulieu, R.; Balicki, D. Aldh1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev. 2009, 18, 17–25. [Google Scholar] [CrossRef]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. Aldh1a1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef]
- Nishida, S.; Hirohashi, Y.; Torigoe, T.; Inoue, R.; Kitamura, H.; Tanaka, T.; Takahashi, A.; Asanuma, H.; Masumori, N.; Tsukamoto, T.; et al. Prostate cancer stem-like cells/cancer-initiating cells have an autocrine system of hepatocyte growth factor. Cancer Sci 2013, 104, 431–436. [Google Scholar] [CrossRef]
- Sefah, K.; Bae, K.M.; Phillips, J.A.; Siemann, D.W.; Su, Z.; McClellan, S.; Vieweg, J.; Tan, W. Cell-based selection provides novel molecular probes for cancer stem cells. Int. J. Cancer 2013, 132, 2578–2588. [Google Scholar] [CrossRef]
- Le Magnen, C.; Bubendorf, L.; Rentsch, C.A.; Mengus, C.; Gsponer, J.; Zellweger, T.; Rieken, M.; Thalmann, G.N.; Cecchini, M.G.; Germann, M.; et al. Characterization and clinical relevance of aldhbright populations in prostate cancer. Clin. Cancer Res. 2013, 19, 5361–5371. [Google Scholar] [CrossRef]
- Yan, J.; De Melo, J.; Cutz, J.C.; Aziz, T.; Tang, D. Aldehyde dehydrogenase 3a1 associates with prostate tumorigenesis. Br. J. Cancer 2014, 110, 2593–2603. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Greten, F.R. Cancer: Tumour stem-cell surprises. Nature 2017, 543, 626–627. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef]
- Naik, S.; Larsen, S.B.; Cowley, C.J.; Fuchs, E. Two to tango: Dialog between immunity and stem cells in health and disease. Cell 2018, 175, 908–920. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, D.G.; Rycaj, K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin. Cancer Biol. 2018, 52, 94–106. [Google Scholar] [CrossRef]
- Pazhanisamy, S.K. Stem cells, DNA damage, ageing and cancer. Hematol./Oncol. Stem Cell Ther. 2009, 2, 375–384. [Google Scholar] [CrossRef]
- Lin, X.; Yan, J.; Tang, D. Erk kinases modulate the activation of pi3 kinase related kinases (pikks) in DNA damage response. Histol. Histopathol. 2013, 28, 1547–1554. [Google Scholar] [PubMed]
- Wei, F.; Yan, J.; Tang, D. Extracellular signal-regulated kinases modulate DNA damage response—a contributing factor to using mek inhibitors in cancer therapy. Curr. Med. Chem. 2011, 18, 5476–5482. [Google Scholar] [CrossRef]
- Burkhalter, M.D.; Rudolph, K.L.; Sperka, T. Genome instability of ageing stem cells--induction and defence mechanisms. Ageing Res. Rev. 2015, 23, 29–36. [Google Scholar] [CrossRef]
- Fan, C.; Quan, R.; Feng, X.; Gillis, A.; He, L.; Matsumoto, E.D.; Salama, S.; Cutz, J.C.; Kapoor, A.; Tang, D. Atm activation is accompanied with earlier stages of prostate tumorigenesis. Biochim. Biophys. Acta 2006, 1763, 1090–1097. [Google Scholar] [CrossRef]
- Park, I.K.; Morrison, S.J.; Clarke, M.F. Bmi1, stem cells, and senescence regulation. J. Clin. Investig. 2004, 113, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16INK4a and p19ARF senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef]
- Bruggeman, S.W.; Valk-Lingbeek, M.E.; van der Stoop, P.P.; Jacobs, J.J.; Kieboom, K.; Tanger, E.; Hulsman, D.; Leung, C.; Arsenijevic, Y.; Marino, S.; et al. INK4a and ARF differentially affect cell proliferation and neural stem cell self-renewal in bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef]
- Oguro, H.; Iwama, A.; Morita, Y.; Kamijo, T.; van Lohuizen, M.; Nakauchi, H. Differential impact of ink4a and arf on hematopoietic stem cells and their bone marrow microenvironment in bmi1-deficient mice. J. Exp. Med. 2006, 203, 2247–2253. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The intestinal stem cell markers bmi1 and lgr5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef]
- Siddique, H.R.; Saleem, M. Role of bmi1, a stem cell factor, in cancer recurrence and chemoresistance: Preclinical and clinical evidences. Stem Cells 2012, 30, 372–378. [Google Scholar] [CrossRef]
- Lin, X.; Gu, Y.; Tang, D. Bmi1, atm and ddr. Oncoscience 2015, 2, 665–666. [Google Scholar]
- Lin, X.; Wei, F.; Whyte, P.; Tang, D. Bmi1 reduces atr activation and signalling caused by hydroxyurea. Oncotarget 2017, 8, 89707–89721. [Google Scholar] [CrossRef]
- Wei, F.; Ojo, D.; Lin, X.; Wong, N.; He, L.; Yan, J.; Xu, S.; Major, P.; Tang, D. Bmi1 attenuates etoposide-induced g2/m checkpoints via reducing atm activation. Oncogene 2015, 34, 3063–3075. [Google Scholar] [CrossRef]
- Yan, J.; Tang, D. Prostate cancer stem-like cells proliferate slowly and resist etoposide-induced cytotoxicity via enhancing DNA damage response. Exp. Cell Res. 2014, 328, 132–142. [Google Scholar] [CrossRef]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and cancer stem cells: Unleashing, hijacking, and restricting cellular plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef]
- Simon, J.A.; Lange, C.A. Roles of the ezh2 histone methyltransferase in cancer epigenetics. Mutat Res 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; Le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. Ezh2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, C.; Zhou, B.; Bi, L.; Huang, H.; Lin, T.; Xu, K. Role of ezh2 in the growth of prostate cancer stem cells isolated from lncap cells. Int. J. Mol. Sci. 2013, 14, 11981–11993. [Google Scholar] [CrossRef]
- Liu, R.; Liu, C.; Zhang, D.; Liu, B.; Chen, X.; Rycaj, K.; Jeter, C.; Calhoun-Davis, T.; Li, Y.; Yang, T.; et al. Mir-199a-3p targets stemness-related and mitogenic signaling pathways to suppress the expansion and tumorigenic capabilities of prostate cancer stem cells. Oncotarget 2016, 7, 56628–56642. [Google Scholar] [CrossRef]
- Chakraborty, C.; Chin, K.Y.; Das, S. Mirna-regulated cancer stem cells: Understanding the property and the role of mirna in carcinogenesis. Tumour. Biol. 2016, 37, 13039–13048. [Google Scholar] [CrossRef]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Sannino, G.; Marchetto, A.; Kirchner, T.; Grunewald, T.G.P. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transition in mesenchymal tumors: A paradox in sarcomas? Cancer Res. 2017, 77, 4556–4561. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Ma, Z.; Yeh, C.R.; Luo, J.; Lin, T.H.; Lai, K.P.; Yamashita, S.; Liang, L.; Tian, J.; Li, L.; et al. New therapy targeting differential androgen receptor signaling in prostate cancer stem/progenitor vs. Non-stem/progenitor cells. J. Mol. Cell Biol. 2013, 5, 14–26. [Google Scholar] [CrossRef]
- Lin, Y.C.; Murayama, Y.; Hashimoto, K.; Nakamura, Y.; Lin, C.S.; Yokoyama, K.K.; Saito, S. Role of tumor suppressor genes in the cancer-associated reprogramming of human induced pluripotent stem cells. Stem Cell Res 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent spop, foxa1 and med12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Blattner, M.; Lee, D.J.; O’Reilly, C.; Park, K.; MacDonald, T.Y.; Khani, F.; Turner, K.R.; Chiu, Y.L.; Wild, P.J.; Dolgalev, I.; et al. Spop mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia 2014, 16, 14–20. [Google Scholar] [CrossRef]
- Boysen, G.; Barbieri, C.E.; Prandi, D.; Blattner, M.; Chae, S.S.; Dahija, A.; Nataraj, S.; Huang, D.; Marotz, C.; Xu, L.; et al. SPOP mutation leads to genomic instability in prostate cancer. eLife 2015, 4. [Google Scholar] [CrossRef]
- Xie, Y.; Zheng, L.; Tao, L. Downregulation of iqgap2 correlates with prostate cancer recurrence and metastasis. Transl. Oncol. 2019, 12, 236–244. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.; Zhu, Y.; Dai, X.; Dang, F.; Ren, J.; Ren, S.; Shulga, Y.V.; Beca, F.; Gan, W.; et al. SPOP promotes NANOG destruction to suppress stem cell traits and prostate cancer progression. Dev. Cell 2019, 48, 329–344.e5. [Google Scholar] [CrossRef]
- Wang, X.; Jin, J.; Wan, F.; Zhao, L.; Chu, H.; Chen, C.; Liao, G.; Liu, J.; Yu, Y.; Teng, H.; et al. AMPK promotes Spop-mediated NANOG degradation to regulate prostate cancer cell stemness. Dev. Cell 2018. [Google Scholar] [CrossRef]
- Lin, C.J.; Lo, U.G.; Hsieh, J.T. The regulatory pathways leading to stem-like cells underlie prostate cancer progression. Asian J. Androl. 2019, 48, 345–360.e7. [Google Scholar]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef]
- Akamatsu, S.; Inoue, T.; Ogawa, O.; Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. 2018, 25, 345–351. [Google Scholar] [CrossRef]
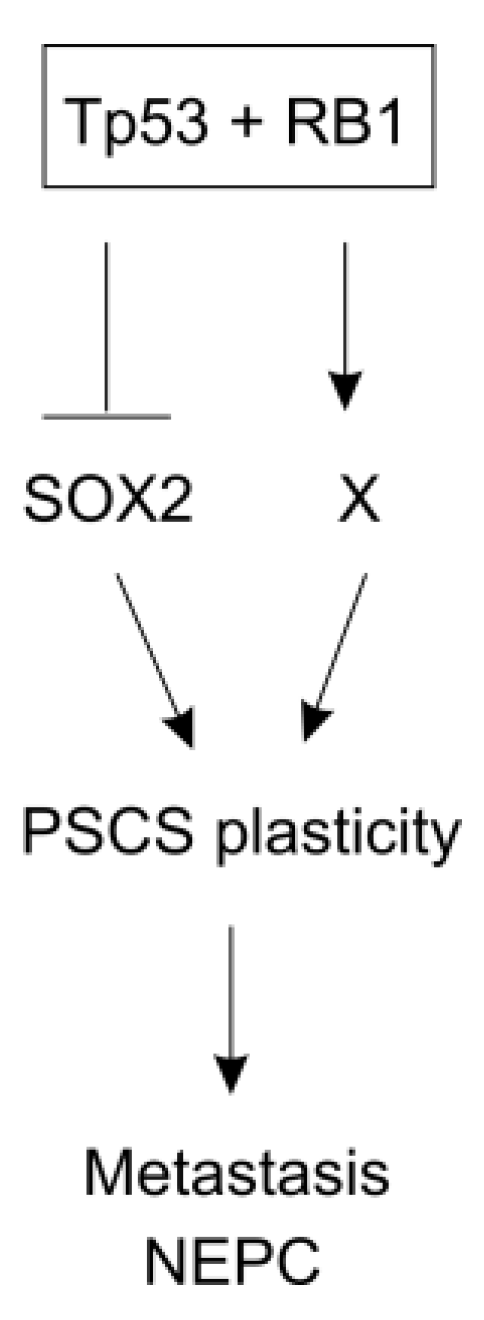
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Soundararajan, R.; Aparicio, A.M.; Logothetis, C.J.; Mani, S.A.; Maity, S.N. Function of tumor suppressors in resistance to antiandrogen therapy and luminal epithelial plasticity of aggressive variant neuroendocrine prostate cancers. Front. Oncol. 2018, 8, 69. [Google Scholar] [CrossRef]
- Soundararajan, R.; Paranjape, A.N.; Maity, S.; Aparicio, A.; Mani, S.A. Emt, stemness and tumor plasticity in aggressive variant neuroendocrine prostate cancers. Biochim. Biophys. Acta. Rev. Cancer 2018, 1870, 229–238. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef]
- Sarig, R.; Rivlin, N.; Brosh, R.; Bornstein, C.; Kamer, I.; Ezra, O.; Molchadsky, A.; Goldfinger, N.; Brenner, O.; Rotter, V. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J. Exp. Med. 2010, 207, 2127–2140. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. Sox2 promotes lineage plasticity and antiandrogen resistance in tp53- and rb1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Karthaus, W.R.; Sawyers, C.L. Strategies to identify and target cells of origin in prostate cancer. J. Natl. Cancer Inst. 2019, 111, 221–223. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Species | PSC 1 | Oncogenic Signal 2 | Tumor Model | Ref. |
|---|---|---|---|---|
| Human | Basal CD49fhiTrop2hi | AKT–ERG–AR | NSG s.c.—xenograft | [50] |
| Mouse | Basal Lin−Sca-1+CD49fhi | AKT1–AR | CB.17SCID/SCID renal capsule | [55] |
| Mouse | Basal Lin−Sca-1+CD49fhi | PTEN knockout | SCID s.c.—xenograft | [56] |
| Mouse | Basal CK5+ | PTEN knockout | Lineage-tracing | [60] |
| Mouse | Basal CK14+ | PTEN knockout | Lineage-tracing | [63] |
| Mouse | Luminal CK8+ | PTEN knockout | Lineage-tracing | [63] |
| Mouse | Luminal Nkx3.1expression | PTEN knockoutNkx3.1 knockout | Lineage-tracing 3 | [65] |
| Mouse | Luminal BMI1+ | PTEN knockout | Lineage-tracing | [67] |
| Mouse | Luminal LY6D+ | PTEN knockout | Lineage-tracing—PIN lesion 4 | [72] |
| Tumor 1 | CTC 2 | Outcome 3 | Metastasis 4 | Ref. |
|---|---|---|---|---|
| BC | EpCAM+CD44+CD47+MET+ | OS 6 | NSG mice; bone mets 5 | [126] |
| BC | EpCAM−HER2+EGFR+HPSE+NOTCH1+ | NA 7 | Nude mice; lung met 8, brain met 9 | [127] |
| CRC | CTC lines with CSC properties | NA 7 | Nude mice; lung and liver met 10 | [128] |
| PC | CK+Vimentin+CD45− | Met 11 | NA 7 | [100] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers 2019, 11, 434. https://doi.org/10.3390/cancers11040434
Mei W, Lin X, Kapoor A, Gu Y, Zhao K, Tang D. The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers. 2019; 11(4):434. https://doi.org/10.3390/cancers11040434
Chicago/Turabian StyleMei, Wenjuan, Xiaozeng Lin, Anil Kapoor, Yan Gu, Kuncheng Zhao, and Damu Tang. 2019. "The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis" Cancers 11, no. 4: 434. https://doi.org/10.3390/cancers11040434
APA StyleMei, W., Lin, X., Kapoor, A., Gu, Y., Zhao, K., & Tang, D. (2019). The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers, 11(4), 434. https://doi.org/10.3390/cancers11040434