Impact of Curcumin (with or without Piperine) on the Pharmacokinetics of Tamoxifen

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Pharmacokinetics
2.3. Toxicities
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Study Procedures
4.3. Pharmacokinetic Sampling
4.4. Toxicity
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative, G. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar]
- Damery, S.; Gratus, C.; Grieve, R.; Warmington, S.; Jones, J.; Routledge, P.; Greenfield, S.; Dowswell, G.; Sherriff, J.; Wilson, S. The use of herbal medicines by people with cancer: A cross-sectional survey. Br. J. Cancer 2011, 104, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.A.; Gescher, A.J.; Steward, W.P. Curcumin: The story so far. Eur. J. Cancer 2005, 41, 1955–1968. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Binkhorst, L.; Mathijssen, R.H.; Jager, A.; van Gelder, T. Individualization of tamoxifen therapy: Much more than just cyp2d6 genotyping. Cancer Treat. Rev. 2015, 41, 289–299. [Google Scholar] [CrossRef]
- Del Re, M.; Citi, V.; Crucitta, S.; Rofi, E.; Belcari, F.; van Schaik, R.H.; Danesi, R. Pharmacogenetics of cyp2d6 and tamoxifen therapy: Light at the end of the tunnel? Pharmacol. Res. 2016, 107, 398–406. [Google Scholar] [CrossRef]
- Cho, Y.A.; Lee, W.; Choi, J.S. Effects of curcumin on the pharmacokinetics of tamoxifen and its active metabolite, 4-hydroxytamoxifen, in rats: Possible role of cyp3a4 and p-glycoprotein inhibition by curcumin. Pharmazie 2012, 67, 124–130. [Google Scholar]
- Bahramsoltani, R.; Rahimi, R.; Farzaei, M.H. Pharmacokinetic interactions of curcuminoids with conventional drugs: A review. J. Ethnopharmacol. 2017, 209, 1–12. [Google Scholar] [CrossRef]
- Ben-Arye, E.; Samuels, N.; Goldstein, L.H.; Mutafoglu, K.; Omran, S.; Schiff, E.; Charalambous, H.; Dweikat, T.; Ghrayeb, I.; Bar-Sela, G.; et al. Potential risks associated with traditional herbal medicine use in cancer care: A study of middle eastern oncology health care professionals. Cancer 2016, 122, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Juan, H.; Jing, T.; Wan-Hua, Y.; Juan, S.; Xiao-Lei, L.; Wen-Xing, P. P-gp induction by curcumin: An effective antidotal pathway. J. Bioequivalence Bioavailab. 2013, 5, 236–241. [Google Scholar]
- Antunes, M.V.; de Oliveira, V.; Raymundo, S.; Staudt, D.E.; Gossling, G.; Biazus, J.V.; Cavalheiro, J.A.; Rosa, D.D.; Mathy, G.; Wallemacq, P.; et al. Cyp3a4*22 is related to increased plasma levels of 4-hydroxytamoxifen and partially compensates for reduced cyp2d6 activation of tamoxifen. Pharmacogenomics 2015, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Diekstra, M.H.; Klumpen, H.J.; Lolkema, M.P.; Yu, H.; Kloth, J.S.; Gelderblom, H.; van Schaik, R.H.; Gurney, H.; Swen, J.J.; Huitema, A.D.; et al. Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and su12662. Clin. Pharm. 2014, 96, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Volak, L.P.; Ghirmai, S.; Cashman, J.R.; Court, M.H. Curcuminoids inhibit multiple human cytochromes p450, udp-glucuronosyltransferase, and sulfotransferase enzymes, whereas piperine is a relatively selective cyp3a4 inhibitor. Drug Metab. Dispos. 2008, 36, 1594–1605. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.L.; Takahashi, K.; Tanaka, K.; Tougou, K.; Qiu, F.; Komatsu, K.; Takahashi, K.; Azuma, J. Curcuma drugs and curcumin regulate the expression and function of p-gp in caco-2 cells in completely opposite ways. Int. J. Pharm. 2008, 358, 224–229. [Google Scholar] [CrossRef]
- Jager, R.; Lowery, R.P.; Calvanese, A.V.; Joy, J.M.; Purpura, M.; Wilson, J.M. Comparative absorption of curcumin formulations. Nutr. J. 2014, 13, 11. [Google Scholar] [CrossRef]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase i clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, H.Y.; Back, S.Y.; Han, H.K. Piperine-mediated drug interactions and formulation strategy for piperine: Recent advances and future perspectives. Expert Opin. Drug Metab. Toxicol. 2018, 14, 43–57. [Google Scholar] [CrossRef]
- Bedada, S.K.; Boga, P.K. The influence of piperine on the pharmacokinetics of fexofenadine, a p-glycoprotein substrate, in healthy volunteers. Eur. J. Clin. Pharm. 2017, 73, 343–349. [Google Scholar] [CrossRef]
- Madlensky, L.; Natarajan, L.; Tchu, S.; Pu, M.; Mortimer, J.; Flatt, S.W.; Nikoloff, D.M.; Hillman, G.; Fontecha, M.R.; Lawrence, H.J.; et al. Tamoxifen metabolite concentrations, cyp2d6 genotype, and breast cancer outcomes. Clin. Pharm. 2011, 89, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Fox, P.; Balleine, R.L.; Lee, C.; Gao, B.; Balakrishnar, B.; Menzies, A.M.; Yeap, S.H.; Ali, S.S.; Gebski, V.; Provan, P.; et al. Dose escalation of tamoxifen in patients with low endoxifen level: Evidence for therapeutic drug monitoring-the tade study. Clin. Cancer Res. 2016, 22, 3164–3171. [Google Scholar] [CrossRef] [PubMed]
- Koolen, S.L.; Bins, S.; Mathijssen, R.H. Individualized tamoxifen dose escalation-letter. Clin. Cancer Res. 2016, 22, 6300. [Google Scholar] [CrossRef] [PubMed]
- Binkhorst, L.; Mathijssen, R.H.; Ghobadi Moghaddam-Helmantel, I.M.; de Bruijn, P.; van Gelder, T.; Wiemer, E.A.; Loos, W.J. Quantification of tamoxifen and three of its phase-i metabolites in human plasma by liquid chromatography/triple-quadrupole mass spectrometry. J. Pharm. Biomed. Anal. 2011, 56, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Jager, N.G.; Rosing, H.; Schellens, J.H.; Linn, S.C.; Beijnen, J.H. Tamoxifen dose and serum concentrations of tamoxifen and six of its metabolites in routine clinical outpatient care. Breast Cancer Res. Treat. 2014, 143, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, D. Statistical Considerations for a Cross-Over Study Where the Outcome is a Measurement. Available online: http://hedwig.mgh.harvard.edu/sample_size/js/js_crossover_quant.html (accessed on 12 January 2017).
- Agency, E.M. Guideline on the Investigation of Bioequivalence. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000370.jsp&mid=WC0b01ac0580032ec5 (accessed on 12 January 2017).
- Kenward, M.G.; Jones, B. Design and Analysis of Cross-Over Trials; Chapman&Hall/CRC monographs: Boca Raton, FL, USA, 2003. [Google Scholar]
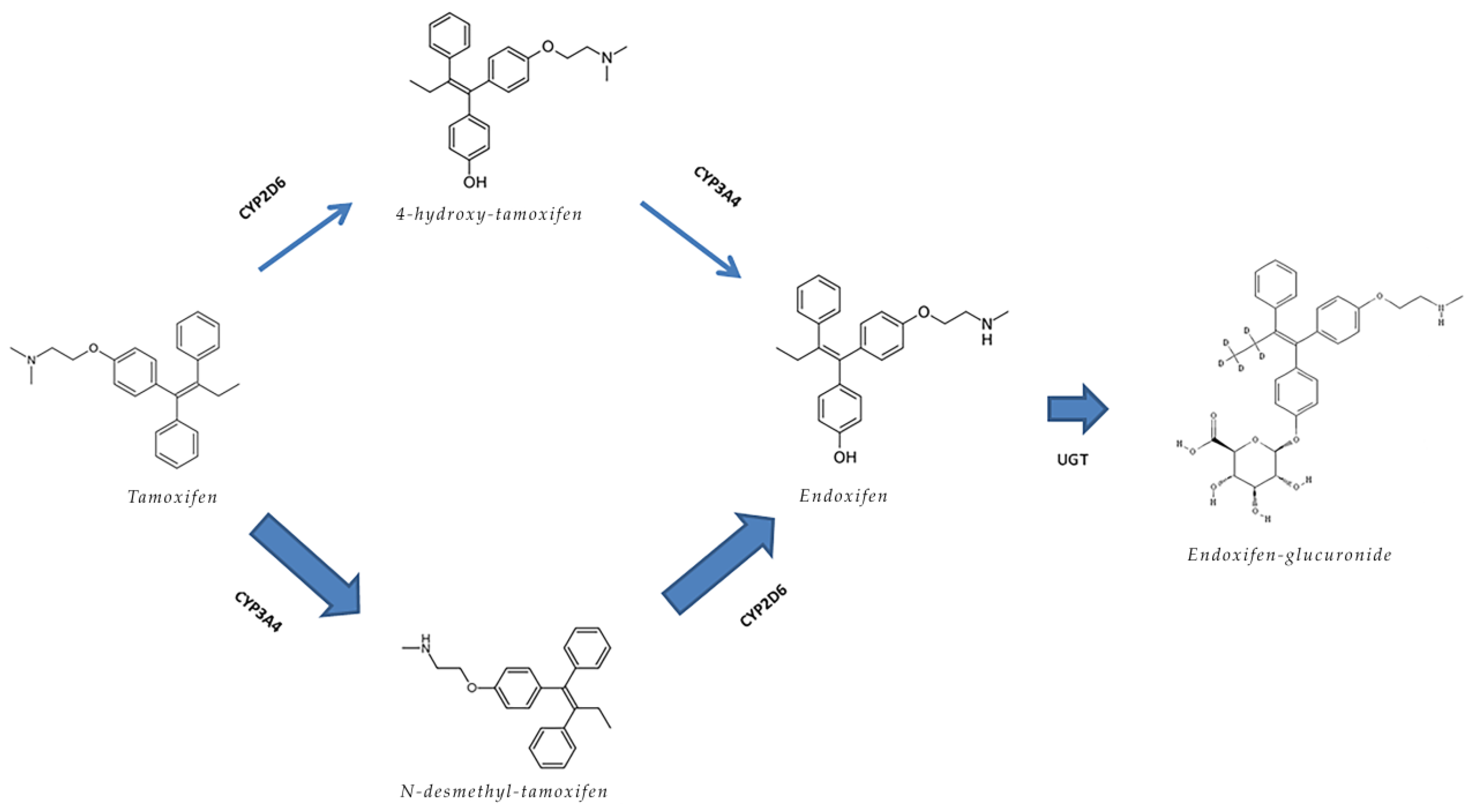
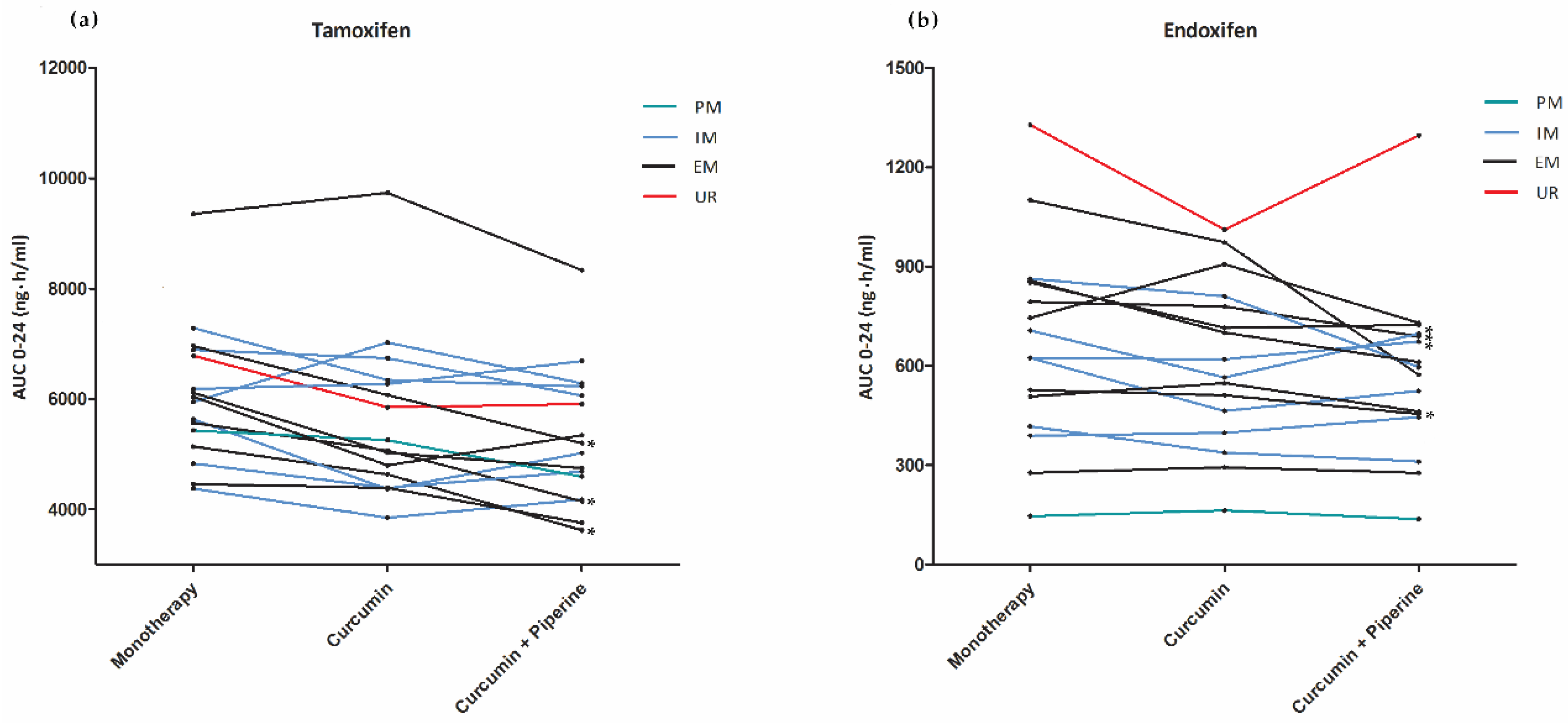

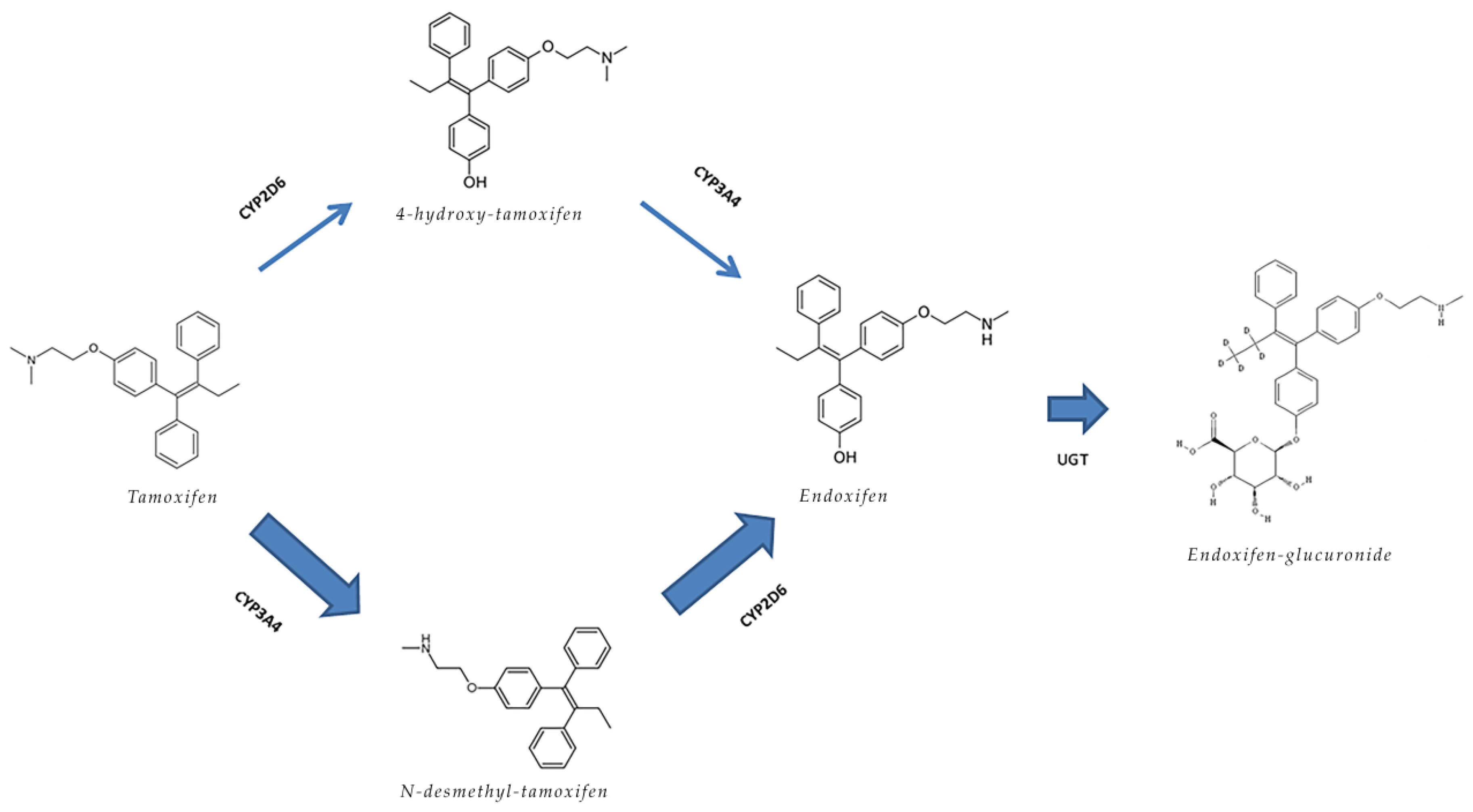{kind=link}
{kind=link}
| Characteristic | N (%) |
|---|---|
| Patients | 16 (100) |
| Randomization sequence | |
| - ABC | 9 (56) |
| - CBA | 7 (44) |
| Adjuvant tamoxifen treatment | 16 (100) |
| Age (Median, IQR) | 45 (42–58) |
| Sex | |
| - Female | 15 (94) |
| - Male | 1 (6) |
| Race | |
| - Caucasian | 15 (94) |
| - Arabic | 1 (6) |
| Height (Median, IQR) | 171 (167–176) |
| Weight (Median, IQR) | 73 (65–91) |
| BMI (Median, IQR) | 25 (23–29) |
| WHO Performance Status | |
| - 0 | 13 (81) |
| - 1 | 3 (19) |
| Previous chemotherapy | |
| - Yes | 12 (75) |
| ○ TAC | 2 (13) |
| ○ AC - paclitaxel | 4 (25) |
| ○ FEC - docetaxel | 6 (37) |
| - No | 4 (25) |
| Previous RTx | |
| - Yes | 10 (63) |
| - No | 6 (37) |
| Tamoxifen dose | |
| - 20 mg | 15 (94) |
| - 30 mg | 1 (6) |
| Genotype | |
| - CYP3A4*22 | |
| ○ EM | 16 (100) |
| - CYP2D6 | |
| ○ EM | 7 (44) |
| ○ IM | 7 (44) |
| ○ PM | 1 (6) |
| ○ UM | 1 (6) |
| PK Parameters | Tamoxifen Monotherapy (A) | Tamoxifen + Curcumin (B) | Tamoxifen + Curcumin + Piperine (C) | Relative Difference A vs B (95%CI) | p-Value | Relative Difference (A vs C) (95%CI) | p-Value |
|---|---|---|---|---|---|---|---|
| Tamoxifen | |||||||
| AUC0–24h | 5951 (20) | 5460 (24) | 5171 (23) | −8.0% (−14.1 to −1.4) | 0.02 | −12.8% (−19.2 to −5.9) | <0.01 |
| Ctrough | 213 (27) | 198 (28) | 187 (24) | −7.1% (−17.1 to +4.0) | 0.25 | −12.2% (−21.5 to −1.8) | 0.02 |
| Cmax | 356 (16) | 324 (21) | 313 (22) | −8.4% (−16.4 to +0.5) | 0.07 | −11.1% (−18.1 to −3.6) | <0.01 |
| Tmax | 2.4 (1.9 to 3.1) | 2.4 (1.9 to 3.0) | 2.7 (1.9 to 3.8) | 0.74 | 0.34 | ||
| Endoxifen | |||||||
| AUC0–24h | 597 (59) | 556 (52) | 518 (54) | −7.7 % (−15.4 to +0.7) | 0.07 | −12.4% (−21.9 to −1.9) | 0.02 |
| Ctrough | 25 (60) | 23 (53) | 21 (55) | −5.6 % (−15.6 to +5.5) | 0.43 | −12.4% (−20.9 to −3.0) | 0.01 |
| Cmax | 31 (56) | 28 (50) | 27 (51) | −7.1% (−16.3 to +3.2) | 0.20 | −9.8% (−20.1 to +1.8) | 0.10 |
| Tmax | 2.0 (1.3 to 3.0) | 1.7 (1.2 to 2.6) | 1.8 (1.1 to 3.1) | 0.88 | 0.62 | ||
| 4-hydroxy-tamoxifen | |||||||
| AUC0–24h | 113 (31) | 106 (24) | 103 (28) | −6.3 % (−11.6 to −0.73) | 0.03 | −8.2% (−17.0 to +1.6) | 0.11 |
| Ctrough | 4.4 (34) | 4.2 (26) | 4.1 (28) | −4.3 % (−12.3 to +4.4) | 0.45 | −7.3 (−17.6 to +4.3) | 0.26 |
| Cmax | 6.0 (32) | 5.4 (26) | 5.4 (31) | −10.0% (−16.8 to −2.6) | <0.01 | −8.8% (−20.0 to +4.0) | 0.20 |
| Tmax | 2.7 (1.9 to 3.9) | 2.4 (1.8 to 3.3) | 2.8 (2.0 to 3.8) | 0.42 | 0.37 | ||
| N-desmethyl-tamoxifen | |||||||
| AUC0–24h | 11596 (21) | 10766 (24) | 10084 (31) | −7.0% (−13.1 to −0.6) | 0.03 | −12.4% (−22.3 to −1.3) | 0.03 |
| Ctrough | 463 (28) | 430 (29) | 411 (32) | −7.2% (−15.0 to +1.2) | 0.10 | −10.9% (−21.6 to +1.3) | 0.08 |
| Cmax | 602 (21) | 556 (24) | 540 (32) | −7.2% (−14.9 to +1.1) | 0.09 | −9.7% (−20.2 to +2.3) | 0.12 |
| Tmax | 2.6 (1.8 to 3.7) | 1.7 (1.2 to 2.4) | 2.1 (1.4 to 3.2) | 0.24 | 0.88 | ||
| PK Parameters | Tamoxifen Monotherapy (A) | Tamoxifen + Curcumin (B) | Tamoxifen + Curcumin + Piperine (C) | Relative Difference A vs B (95%CI) | p-Value | Relative Difference A vs C (95%CI) | p-Value |
|---|---|---|---|---|---|---|---|
| Intermediate Metabolizers (IM) | |||||||
| Tamoxifen AUC0–24h | 5795 (4895–6859) | 5427 (4313–6830) | 5518 (4679–6508) | −7.2% (−18.2 to +5.4) | 0.19 | −5.3% (−13.1 to +3.1) | 0.16 * |
| Tamoxifen Ctrough | 200 (160–251) | 191 (146–249) | 199 (167–237) | −5.9% (−20.9to +11.9) | 0.41 | −1.3% (−15.3 to 15.1) | 0.84 * |
| Endoxifen AUC0–24h | 523 (362–755) | 472 (339–656) | 477 (340–669) | −9.4% (−21.7 to +4.8) | 0.14 | −10.3% (−23.5 to 5.3) | 0.14 |
| Endoxifen Ctrough | 21 (14–32) | 19 (13–27) | 19 (14–27) | −10.7% (−28.2 to 11.2) | 0.24 | −8.3% (−27.2 to 15.4) | 0.38 |
| Extensive Metabolizers (EM) | |||||||
| Tamoxifen AUC0–24h | 6077 (4882–7565) | 5471 (4247–7047) | 4836 (3720–6288) | −10.3% (−19.7 to +0.3) | 0.06 | −22.0% (−29.0 to −4.2) | <0.01 * |
| Tamoxifen Ctrough | 218 (163–291) | 199 (148–268) | 170 (132–218) | −9.6% (−26.4 to +11.2) | 0.27 | −24.6% (−33.9 to −14.1) | <0.01 * |
| Endoxifen AUC0–24h | 745 (576–963) | 716 (574–893) | 596 (495–717) | −5.7% (−19.6 to +10.7) | 0.39 | −18.4% (−36.1 to +4.3) | 0.09 |
| Endoxifen Ctrough | 30 (23–39) | 31 (25–38) | 25 (20–30) | −0.3% (−12.8 to +13.9) | 0.96 | −17.2% (−26.1 to −7.3) | <0.01 * |
| Adverse Event | Tamoxifen Monotherapy N (%) (A) | Tamoxifen with Curcumin N (%) (B) | Tamoxifen with Curcumin and Piperine N (%) (C) |
|---|---|---|---|
| Nausea | 2(13) | 1(6) | 1(6) |
| Diarrhea | 0 | 1(6) | 3(19) |
| Constipation | 2(13) | 4(25) | 1(6) |
| Fatigue | 2(13) | 3(19) | 3(19) |
| Hot flashes | 3(19) | 5(31) | 4(25) |
| Reflux | 1(6) | 1(6) | 0 |
| Dyspnea | 0 | 1(6) | 0 |
| Anorexia | 1(6) | 0 | 1(6) |
| Pain | 4(25) | 0 | 2(13) |
| Rash | 1(6) | 0 | 1(6) |
| Hypophosphatemia | 0 | 0 | 1(6) |
| Hyperlipidemia | 1(6) | 1(6) | 1(6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussaarts, K.G.A.M.; Hurkmans, D.P.; Oomen-de Hoop, E.; van Harten, L.J.; Berghuis, S.; van Alphen, R.J.; Spierings, L.E.A.; van Rossum-Schornagel, Q.C.; Vastbinder, M.B.; van Schaik, R.H.N.; et al. Impact of Curcumin (with or without Piperine) on the Pharmacokinetics of Tamoxifen. Cancers 2019, 11, 403. https://doi.org/10.3390/cancers11030403
Hussaarts KGAM, Hurkmans DP, Oomen-de Hoop E, van Harten LJ, Berghuis S, van Alphen RJ, Spierings LEA, van Rossum-Schornagel QC, Vastbinder MB, van Schaik RHN, et al. Impact of Curcumin (with or without Piperine) on the Pharmacokinetics of Tamoxifen. Cancers. 2019; 11(3):403. https://doi.org/10.3390/cancers11030403
Chicago/Turabian StyleHussaarts, Koen G.A.M., Daan P. Hurkmans, Esther Oomen-de Hoop, Leonie J. van Harten, Stan Berghuis, Robbert J. van Alphen, Leontine E.A. Spierings, Quirine C. van Rossum-Schornagel, Mijntje B. Vastbinder, Ron H.N. van Schaik, and et al. 2019. "Impact of Curcumin (with or without Piperine) on the Pharmacokinetics of Tamoxifen" Cancers 11, no. 3: 403. https://doi.org/10.3390/cancers11030403
APA StyleHussaarts, K. G. A. M., Hurkmans, D. P., Oomen-de Hoop, E., van Harten, L. J., Berghuis, S., van Alphen, R. J., Spierings, L. E. A., van Rossum-Schornagel, Q. C., Vastbinder, M. B., van Schaik, R. H. N., van Gelder, T., Jager, A., van Leeuwen, R. W. F., & Mathijssen, R. H. J. (2019). Impact of Curcumin (with or without Piperine) on the Pharmacokinetics of Tamoxifen. Cancers, 11(3), 403. https://doi.org/10.3390/cancers11030403