Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer

 ,
,  , , ,
, , ,  , ,
, , 
 ,
,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Two-Hit Prioritization Strategy Identified New Candidate Genes for CRC Germline Predisposition
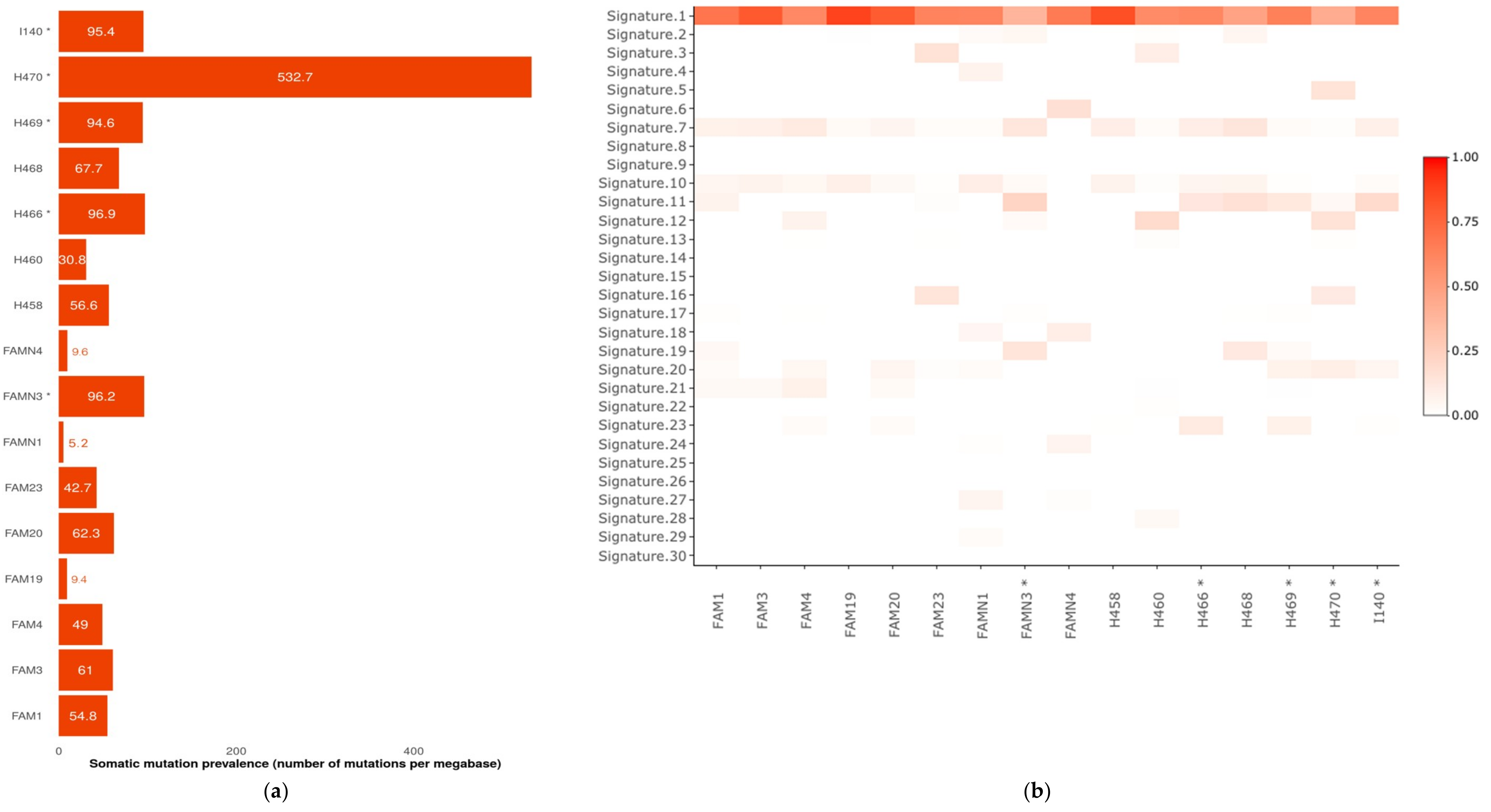
2.2. Somatic Mutational Profiling Detected Hypermutated Tumors Compatible with A Germline Defect Etiology
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole Exome Sequencing
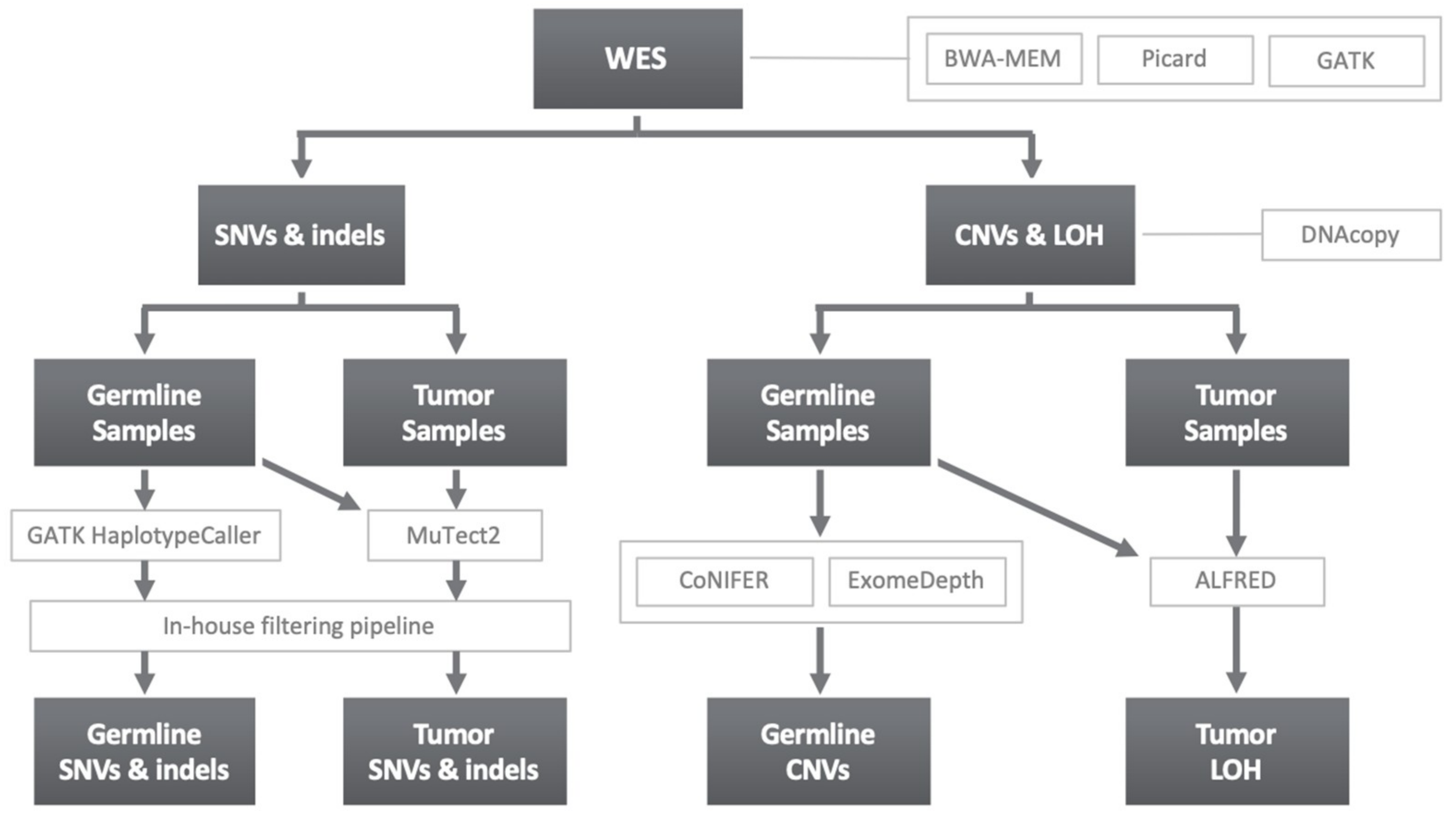
4.3. Variant Calling and Filtering
4.3.1. SNVs and Indels
4.3.2. Copy Number Variants and Loss of Heterozygosity
4.4. Variant Prioritization and Validation
4.5. Mutational Profiling and Mutational Signature Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.K.; Colditz, G.A.; Giovannucci, E.L.; Wu, K.; Glynn, R.J.; Fuchs, C.S.; Stampfer, M.; Willett, W.; Ogino, S.; Rosner, B. A Comprehensive Model of Colorectal Cancer by Risk Factor Status and Subsite Using Data From the Nurses’ Health Study. Am. J. Epidemiol. 2017, 185, 224–237. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I. The Mendelian colorectal cancer syndromes. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 2015, 53, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Valle, L. Recent Discoveries in the Genetics of Familial Colorectal Cancer and Polyposis. Clin. Gastroenterol. Hepatol. 2017, 15, 809–819. [Google Scholar] [CrossRef]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Tan, R.; Wang, Y.; Kleinstein, S.E.; Liu, Y.; Zhu, X.; Guo, H.; Jiang, Q.; Allen, A.S.; Zhu, M. An Evaluation of Copy Number Variation Detection Tools from Whole-Exome Sequencing Data. Hum. Mutat. 2014, 35, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Franch-Expósito, S.; Esteban-Jurado, C.; Garre, P.; Quintanilla, I.; Duran-Sanchon, S.; Díaz-Gay, M.; Bonjoch, L.; Cuatrecasas, M.; Samper, E.; Muñoz, J.; et al. Rare germline copy number variants in colorectal cancer predisposition characterized by exome sequencing analysis. J. Genet. Genom. 2018, 45, 41–45. [Google Scholar] [CrossRef]
- Park, S.; Supek, F.; Lehner, B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat. Commun. 2018, 9, 2601. [Google Scholar] [CrossRef] [PubMed]
- Finlin, B.S.; Gau, C.-L.; Murphy, G.A.; Shao, H.; Kimel, T.; Seitz, R.S.; Chiu, Y.-F.; Botstein, D.; Brown, P.O.; Der, C.J.; et al. RERG Is a Novel ras-related, Estrogen-regulated and Growth-inhibitory Gene in Breast Cancer. J. Biol. Chem. 2001, 276, 42259–42267. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Goh, S.-H.; Lee, S.J.; Hwang, J.-A.; Lee, J.; Choi, I.-J.; Seo, H.; Park, J.-H.; Suzuki, H.; Yamamoto, E.; et al. Upregulation of adenylate cyclase 3 (ADCY3) increases the tumorigenic potential of cells by activating the CREB pathway. Oncotarget 2013, 4, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Sun, T.; Luo, J.; McKinstry, R.C.; Parkin, P.C.; Ganzhorn, S.; Spoljaric, D.; Albers, A.C.; Merkelson, A.; Stewart, D.R.; et al. The Cyclic AMP Pathway Is a Sex-Specific Modifier of Glioma Risk in Type I Neurofibromatosis Patients. Cancer Res. 2015, 75, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Handler, M.; Eichstetter, I.; Whitelock, J.M.; Nugent, M.A.; Iozzo, R. V Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J. Clin. Investig. 1998, 102, 1599–1608. [Google Scholar] [CrossRef]
- Sharma, S.; Doherty, K.M.; Brosh, R.M. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem. J. 2006, 398, 319–337. [Google Scholar] [CrossRef]
- Cybulski, C.; Carrot-Zhang, J.; Kluźniak, W.; Rivera, B.; Kashyap, A.; Wokołorczyk, D.; Giroux, S.; Nadaf, J.; Hamel, N.; Zhang, S.; et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet. 2015, 47, 643. [Google Scholar] [CrossRef]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef]
- Esteban-Jurado, C.; Franch-Expósito, S.; Muñoz, J.; Ocaña, T.; Carballal, S.; López-Cerón, M.; Cuatrecasas, M.; Vila-Casadesús, M.; Lozano, J.J.; Serra, E.; et al. The Fanconi anemia DNA damage repair pathway in the spotlight for germline predisposition to colorectal cancer. Eur. J. Hum. Genet. 2016, 24, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- García, M.J.; Fernández, V.; Osorio, A.; Barroso, A.; Llort, G.; Lázaro, C.; Blanco, I.; Caldés, T.; de la Hoya, M.; Ramón y Cajal, T.; et al. Analysis of FANCB and FANCN/PALB2 fanconi anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res. Treat. 2009, 113, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef] [PubMed]
- Coin, F.; Marinoni, J.-C.; Rodolfo, C.; Fribourg, S.; Pedrini, A.M.; Egly, J.-M. Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat. Genet. 1998, 20, 184. [Google Scholar] [CrossRef] [PubMed]
- Frederick, G.D.; Amirkhan, R.H.; Schultz, R.A.; Friedberg, E.C. Structural and mutational analysis of the xeroderma pigmentosum group D (XPD) gene. Hum. Mol. Genet. 1994, 3, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Rump, A.; Benet-Pages, A.; Schubert, S.; Kuhlmann, J.D.; Janavičius, R.; Macháčková, E.; Foretová, L.; Kleibl, Z.; Lhota, F.; Zemankova, P.; et al. Identification and Functional Testing of ERCC2 Mutations in a Multi-national Cohort of Patients with Familial Breast- and Ovarian Cancer. PLOS Genet. 2016, 12, e1006248. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Tiao, G.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.D.; et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef]
- Yang, L.; Shi, T.; Liu, F.; Ren, C.; Wang, Z.; Li, Y.; Tu, X.; Yang, G.; Cheng, X. REV3L, a Promising Target in Regulating the Chemosensitivity of Cervical Cancer Cells. PLoS ONE 2015, 10, e0120334. [Google Scholar] [CrossRef]
- Chapman, J.R.; Barral, P.; Vannier, J.-B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Esteban-Jurado, C.; Vila-Casadesús, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Muñoz, J.; Ocaña, T.; Balaguer, F.; López-Cerón, M.; et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 2015, 17, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056e10. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Iolascon, A.; Verissimo, F.; Trede, N.S.; Horsley, W.; Chen, W.; Paw, B.H.; Hopfner, K.-P.; Holzmann, K.; Russo, R.; et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 2009, 41, 936. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Niazi, F.; Ni, Y.; Ngeow, J.; Sankunny, M.; Liu, Z.; Wei, W.; Mester, J.L.; Keri, R.A.; Zhang, B.; et al. Germline Heterozygous Variants in SEC23B Are Associated with Cowden Syndrome and Enriched in Apparently Sporadic Thyroid Cancer. Am. J. Hum. Genet. 2015, 97, 661–676. [Google Scholar] [CrossRef] [PubMed]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.M.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacoke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.P.; Rayter, S.I.; Niederlander, C.; Spicer, J.; Jones, C.M.; Ashworth, A. LIP1, a cytoplasmic protein functionally linked to the Peutz-Jeghers syndrome kinase LKB1. Hum. Mol. Genet. 2001, 10, 2869–2877. [Google Scholar] [CrossRef]
- Walsh, M.F.; Ritter, D.I.; Kesserwan, C.; Sonkin, D.; Chakravarty, D.; Chao, E.; Ghosh, R.; Kemel, Y.; Wu, G.; Lee, K.; et al. Integrating somatic variant data and biomarkers for germline variant classification in cancer predisposition genes. Hum. Mutat. 2018, 39, 1542–1552. [Google Scholar] [CrossRef]
- Spier, I.; Kerick, M.; Drichel, D.; Horpaopan, S.; Altmüller, J.; Laner, A.; Holzapfel, S.; Peters, S.; Adam, R.; Zhao, B.; et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam. Cancer 2016, 281–288. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Deane, N.G.; Zhu, J.; An, H.; Mima, S.; Wang, X.; Padmanabhan, S.; Shi, Z.; Prodduturi, N.; Ciombor, K.K.; et al. Nuclear factor of activated T-cell activity is associated with metastatic capacity in colon cancer. Cancer Res. 2014, 74, 6947–6957. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, B.; Wolfinger, R.D.; Chen, X. An integrated approach for the analysis of biological pathways using mixed models. PLoS Genet. 2008, 4, e1000115. [Google Scholar] [CrossRef]
- Wang, L.; Chen, X.; Wolfinger, R.D.; Franklin, J.L.; Coffey, R.J.; Zhang, B. A unified mixed effects model for gene set analysis of time course microarray experiments. Stat. Appl. Genet. Mol. Biol. 2009, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Shirts, B.H.; Konnick, E.Q.; Upham, S.; Walsh, T.; Ranola, J.M.O.; Jacobson, A.L.; King, M.-C.; Pearlman, R.; Hampel, H.; Pritchard, C.C. Using Somatic Mutations from Tumors to Classify Variants in Mismatch Repair Genes. Am. J. Hum. Genet. 2018, 103, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Seshan, V.E.; Olshen, A. DNAcopy: DNA Copy Number Data Analysis, R package version 1.48.0. Bioconductor; Roswell Park Comprehensive Cancer Center: Buffalo, NY, USA, 2016. [Google Scholar]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Guo, Y.; Li, J.; Li, C.-I.; Long, J.; Samuels, D.C.; Shyr, Y. The effect of strand bias in Illumina short-read sequencing data. BMC Genom. 2012, 13, 666. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Houlston, R.S. CanVar: A resource for sharing germline variation in cancer patients. F1000Research 2016, 5, 2813. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Vila-Casadesús, M.; Franch-Expósito, S.; Hernández-Illán, E.; Lozano, J.J.; Castellví-Bel, S. Mutational Signatures in Cancer (MuSiCa): A web application to implement mutational signatures analysis in cancer samples. BMC Bioinform. 2018, 19, 224. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Family | RefSeq Transcript | Hit | Genetic Variant | Path. Tools | DAMpred | ExAC Freq. | Protein Domain | Protein Function |
|---|---|---|---|---|---|---|---|---|---|
| ADCY8 | FAMN4 | NM_001115.2 | 1st | c.1747G>A p.(Glu583Lys) | 5/6 | − | 21/60,697 | Adenylyl cyclase class-3/4/guanylyl cyclase domain | Biosynthesis of cAMP from ATP |
| 2nd | c.458C>T p.(Ile153Thr) | 4/6 | + | 0/60,706 | Interaction with ORAI1, STIM1, PPP2CA and PPP2R1A | ||||
| HSPG2 | FAM23 | NM_005529.7 | 1st | c.3148G>A p.(Gly1050Ser) | 3/6 | − | 3/60,456 | Laminin IV type A domain | Component of vascular extracellular matrix, regulation of angiogenesis and cell growth |
| 2nd | c.7406C>T p.(Thr2469Met) | 4/6 | − | 0/60,706 | Immunoglobulin-like C2-type domain |
| Gene | Family | RefSeq Transcript | Genetic Variant | Path. Tools | DAMpred | ExAC Freq. | Protein Domain | Protein Function |
|---|---|---|---|---|---|---|---|---|
| BRCA2 | FAM20 | NM_000059.3 | c.4963delT p.(Tyr1655fs*15) | FS | n.a. | 0/60,706 | - | Double-strand break repair via homologous recombination, inherited predisposition to breast and ovarian cancer |
| BLM | FAMN4 | NM_000057.4 | c.2069C>T p.(Pro690Leu) | 6/6 | + | 1/60,570 | Helicase ATP-binding domain | DNA helicase, double-strand break repair via homologous recombination, regulation of cell cycle and apoptosis, DNA replication, telomere maintenance |
| ERCC2 | H458 | NM_000400.3 | c.688G>A p.(Val230Ile) | 4/6 | − | 0/60,706 | Helicase ATP-binding domain | DNA helicase, transcription-coupled nucleotide excision repair, regulation of cell cycle |
| FAT2 | FAMN3 | NM_001447.2 | c.1643T>C p.(Val548Ala) | 5/6 | − | 0/60,706 | Cadherin domain | Regulation of cell proliferation, cell adhesion |
| IGF2R | H466 | NM_000876.3 | c.232G>A p.(Gly78Arg) | 6/6 | + | 1/60,684 | - | Positive regulation of apoptosis |
| LATS2 | H460 | NM_014572.3 | c.337G>A p.(Asp113Asn) | 5/6 | − | 1/56,138 | Ubiquitin-associated domain | Positive regulation of apoptosis, regulation of cell cycle |
| PARP2 | FAM20 | NM_005484.3 | c.910G>C p.(Glu304Gln) | 3/6 | − | 3/60,208 | Poly(ADP-ribose) polymerase (PARP) alpha-helical domain | Base excision repair, extrinsic apoptotic signaling pathway |
| PSMD9 | H469 | NM_002813.6 | c.361A>T p.(Ser121Cys) | 3/6 | − | 30/60,148 | PDZ domain | Subunit of 26S proteasome, regulation of apoptosis and cell cycle, regulation of ubiquitin-protein ligase activity |
| RASSF6 | H460 | NM_201431.2 | c.779C>T p.(Pro260Leu) | 6/6 | − | 53/60,475 | Ras-associating domain | Positive regulation of apoptosis |
| RECQL | H466 | NM_002907.4 | c.221_225delinsAATGT p.(Pro74_Trp75delinsGlnCys) | 6/6 | + | 0/60,706 | - | DNA helicase, double-strand break repair via homologous recombination, DNA replication |
| RERGL | H466 | NM_024730.3 | c.362T>C p.(Val121Ala) | 6/6 | + | 54/60,446 | - | Unknown (closely related to RERG, which functions as a negative regulator of cell growth [14]) |
| REV3L | FAM3 | NM_002912.4 | c.559A>T p.(Arg187Trp) | 5/6 | − | 0/60,706 | Exonuclease domain (family B of DNA polymerases) | DNA repair, translesion DNA synthesis |
| RIF1 | H460 | NM_018151.4 | c.4262G>A p.(Arg1421His) | 4/6 | + | 5/59,938 | - | Double-strand break repair via nonhomologous end joining, telomere maintenance |
| SEC23B | H470 | NM_032985.5 | c.531G>C p.(Glu177Asp) | 4/6 | − | 1/60,706 | Sec23/Sec24 trunk domain | Intracellular protein transport, associated with inherited cancer predisposition Cowden Syndrome |
| SMARCA4 | FAM3 | NM_003072.3 | c.295C>T p.(Arg99Trp) | 5/6 | − | 1/60,196 | - | Regulation of cell growth, regulation of cell cycle, chromatin remodeling |
| STK11IP | H470 | NM_052902.4 | c.1214C>T p.(Pro405Leu) | 5/6 | − | 51/59,930 | - | Interaction with STK11 (serine/threonine kinase activity, negative regulation of cell growth, Peutz-Jeghers CRC predisposition syndrome) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Gay, M.; Franch-Expósito, S.; Arnau-Collell, C.; Park, S.; Supek, F.; Muñoz, J.; Bonjoch, L.; Gratacós-Mulleras, A.; Sánchez-Rojas, P.A.; Esteban-Jurado, C.; et al. Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer. Cancers 2019, 11, 362. https://doi.org/10.3390/cancers11030362
Díaz-Gay M, Franch-Expósito S, Arnau-Collell C, Park S, Supek F, Muñoz J, Bonjoch L, Gratacós-Mulleras A, Sánchez-Rojas PA, Esteban-Jurado C, et al. Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer. Cancers. 2019; 11(3):362. https://doi.org/10.3390/cancers11030362
Chicago/Turabian StyleDíaz-Gay, Marcos, Sebastià Franch-Expósito, Coral Arnau-Collell, Solip Park, Fran Supek, Jenifer Muñoz, Laia Bonjoch, Anna Gratacós-Mulleras, Paula A. Sánchez-Rojas, Clara Esteban-Jurado, and et al. 2019. "Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer" Cancers 11, no. 3: 362. https://doi.org/10.3390/cancers11030362
APA StyleDíaz-Gay, M., Franch-Expósito, S., Arnau-Collell, C., Park, S., Supek, F., Muñoz, J., Bonjoch, L., Gratacós-Mulleras, A., Sánchez-Rojas, P. A., Esteban-Jurado, C., Ocaña, T., Cuatrecasas, M., Vila-Casadesús, M., Lozano, J. J., Parra, G., Laurie, S., Beltran, S., EPICOLON Consortium, Castells, A., ... Castellví-Bel, S. (2019). Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer. Cancers, 11(3), 362. https://doi.org/10.3390/cancers11030362