Exploring the Frequency of Homologous Recombination DNA Repair Dysfunction in Multiple Cancer Types

 ,
,
Abstract
1. Introduction
2. Results
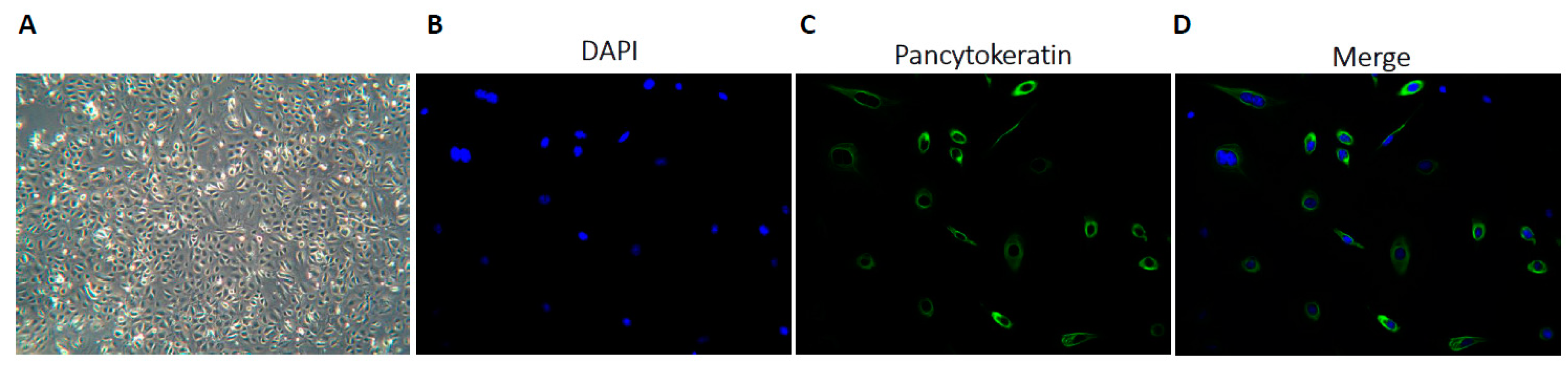
2.1. Establishing Primary Cultures from Ascitic Fluid Samples
2.2. Determining HRR Status in Primary Cultures
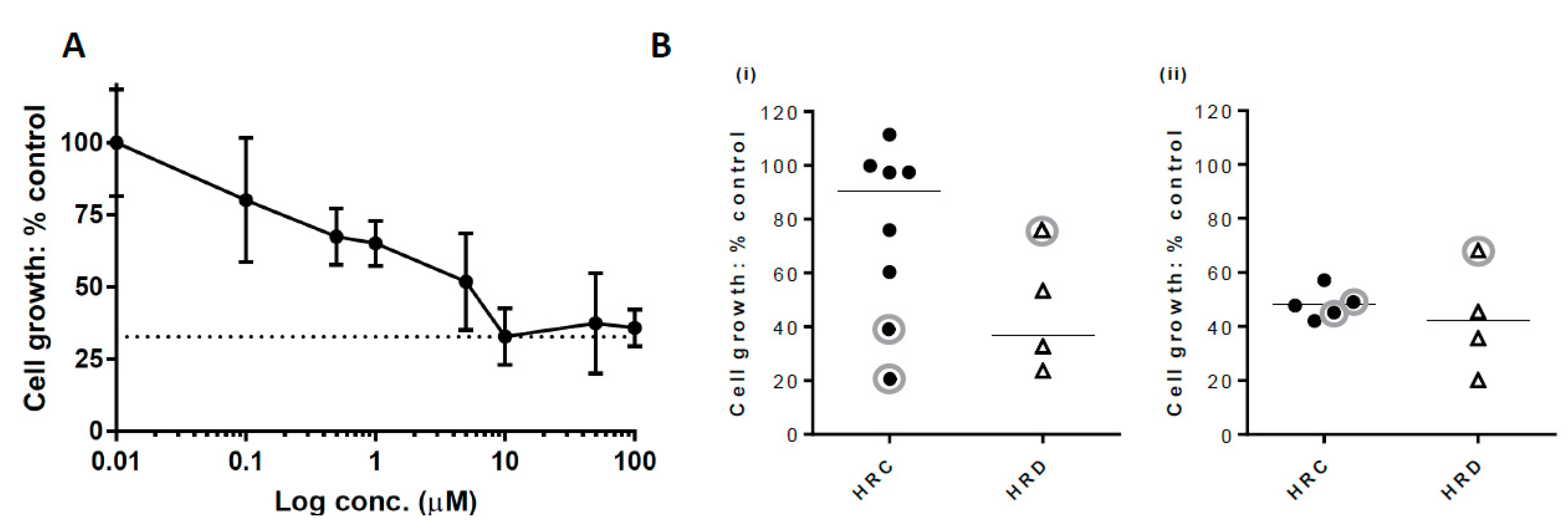
2.3. Growth Inhibition with PARP Inhibitor
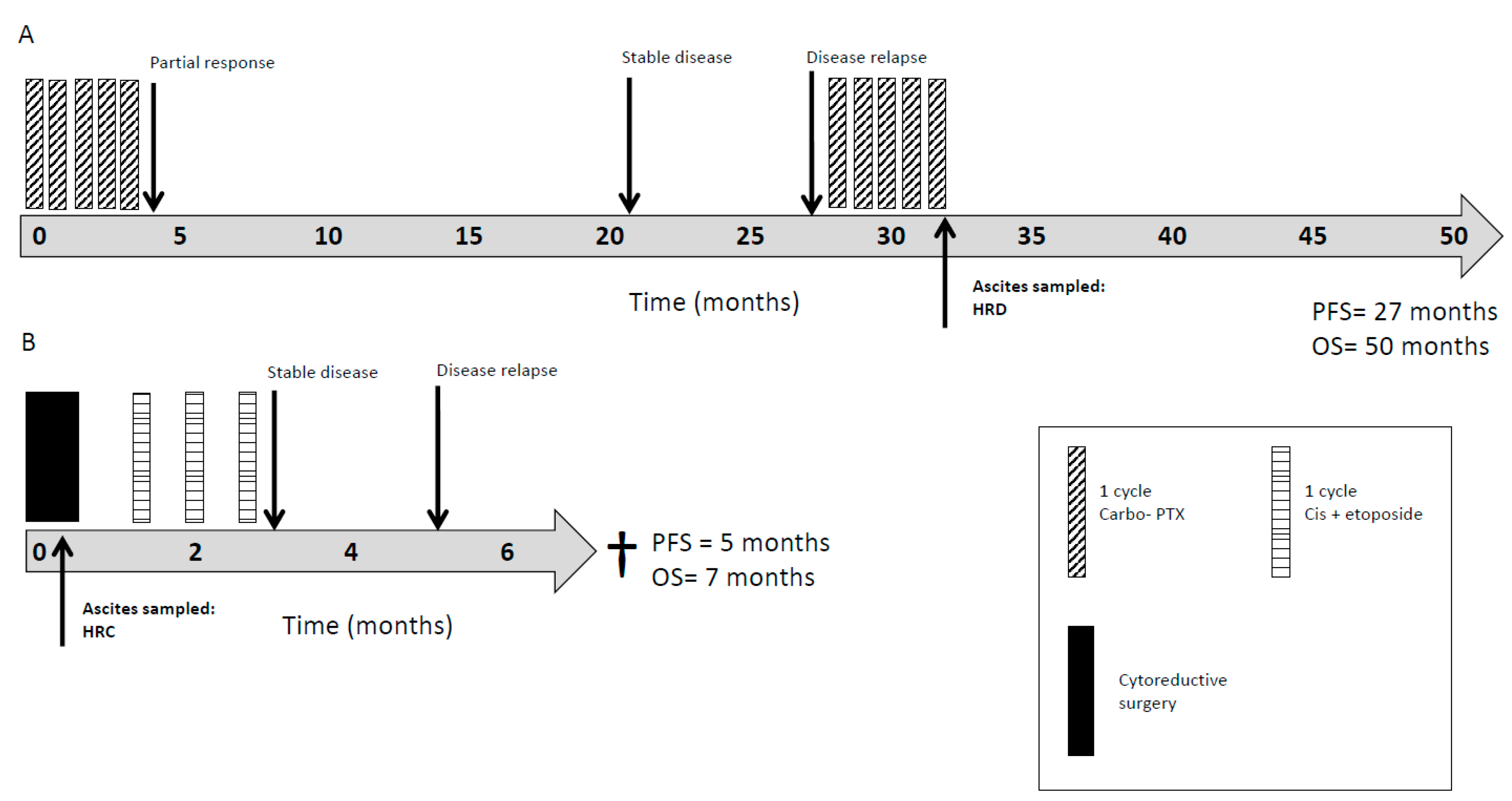
2.4. HRR as a Clinically Prognostic Marker
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Sample Collection and Generation of Primary Cultures
Primary culture
4.3. Immunofluorescent Assays
4.3.1. Epithelial Characterisation
4.3.2. RAD51 Focus Assay for Assessment of HRR Function
4.4. Inhibition of Growth by Rucaparib
4.5. Clinical Response and Survival Data
5. Conclusions
Additional Information
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Elattar, A.; Cerbinskaite, A.; Wilkinson, S.J.; Drew, Y.; Kyle, S.; Los, G.; Hostomsky, Z.; Edmondson, R.J.; Curtin, N.J. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin. Cancer Res. 2010, 16, 2344–2351. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. BCR 2014, 16, 211. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Gheorghiu, L.; Liu, Q.; Efstathiou, J.A.; Wirth, L.J.; Krause, M.; von Neubeck, C. DNA Damage Response Assessments in Human Tumor Samples Provide Functional Biomarkers of Radiosensitivity. Semin. Radiat. Oncol. 2015, 25, 237–250. [Google Scholar] [CrossRef]
- Gry, M.; Rimini, R.; Stromberg, S.; Asplund, A.; Ponten, F.; Uhlen, M.; Nilsson, P. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genom. 2009, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggage, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: Sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012, 72, 5675–5682. [Google Scholar] [CrossRef]
- Patterson, M.J.; Sutton, R.E.; Forrest, I.; Sharrock, R.; Lane, M.; Kaufmann, A.; O’Donnell, R.; Edmondson, R.J.; Wilson, B.T.; Curtin, N.J. Assessing the function of homologous recombination DNA repair in malignant pleural effusion (MPE) samples. Br. J. Cancer 2014, 111, 94–100. [Google Scholar] [CrossRef] [PubMed]
- PARP Inhibitor in Advanced Non-Small Cell Lung (PIN). Available online: https://clinicaltrials.gov/ct2/show/results/NCT01788332 (accessed on 8 March 2019).
- Trial of Gemcitabine/Carboplatin with or without Iniparib (SAR240550) (a PARP1 Inhibitor) in Subjects with Previously Untreated Stage IV Squamous Non-Small Cell Lung (NSCLC) (ECLIPSE). Available online: https://clinicaltrials.gov/ct2/show/NCT01082549 (accessed on 8 March 2019).
- Study of Niraparib Administered Alone and in Combination With PD-1 Inhibitor in Patients with Non-Small Cell Lung. Available online: https://clinicaltrials.gov/ct2/show/NCT03308942 (accessed on 8 March 2019).
- A Study Evaluating the Efficacy and Tolerability of Veliparib in Combination with Paclitaxel/Carboplatin-Based Chemoradiotherapy Followed by Veliparib and Paclitaxel/Carboplatin Consolidation in Subjects with Stage III Non-Small Cell Lung. Available online: https://clinicaltrials.gov/ct2/show/NCT02412371 (accessed on 8 March 2019).
- Study of Veliparib in Combination with Nivolumab and Platinum Doublet Chemotherapy in Participants with Metastatic or Advanced Non-Small Cell Lung (NSCLC). Available online: https://clinicaltrials.gov/ct2/show/NCT02944396 (accessed on 8 March 2019).
- Randomized, Double-Blind, Multicenter, Study Comparing Veliparib Plus Carboplatin and Paclitaxel Versus Placebo Plus Carboplatin and Paclitaxel in Previously Untreated Advanced or Metastatic Squamous Non-Small Cell Lung. Available online: https://clinicaltrials.gov/ct2/show/NCT02106546 (accessed on 8 March 2019).
- Veliparib with or without Radiation Therapy, Carboplatin, and Paclitaxel in Patients with Stage III Non-Small Cell Lung That Cannot Be Removed by Surgery. Available online: https://clinicaltrials.gov/ct2/show/NCT01386385 (accessed on 8 March 2019).
- Study Comparing Veliparib Plus Carboplatin and Paclitaxel Versus Investigator’s Choice of Standard Chemotherapy in Subjects Receiving First Cytotoxic Chemotherapy for Metastatic or Advanced Non-Squamous Non-Small Cell Lung (NSCLC) and Who Are Current or Former Smokers. Available online: https://clinicaltrials.gov/ct2/show/NCT02264990 (accessed on 8 March 2019).
- Cisplatin and Etoposide with or without Veliparib in Treating Patients with Extensive Stage Small Cell Lung or Metastatic Large Cell Neuroendocrine Non-Small Cell Lung. Available online: https://clinicaltrials.gov/ct2/show/NCT01642251 (accessed on 8 March 2019).
- Trial of CRLX101, a Nanoparticle Camptothecin With Olaparib in People with Relapsed/Refractory Small Cell Lung. Available online: https://clinicaltrials.gov/ct2/show/NCT02769962 (accessed on 8 March 2019).
- Olaparib Dose Escalating Trial + Concurrent RT With or Without Cisplatin in Locally Advanced NSCLC (olaparib). Available online: https://clinicaltrials.gov/ct2/show/NCT01562210 (accessed on 8 March 2019).
- A Phase 2 Study of Cediranib in Combination with Olaparib in Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02498613 (accessed on 8 March 2019).
- Novello, S.; Besse, B.; Felip, E.; Barlesi, F.; Mazieres, J.; Zalcman G von Pawel, J.; Reck, M.; Cappuzzo, F.; Ferry, D.; et al. A phase II randomized study evaluating the addition of iniparib to gemcitabine plus cisplatin as first-line therapy for metastatic non-small-cell lung cancer. Ann. Oncol. 2014, 25, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Blais, N.; Mazieres, J.; Reck, M.; Jones, C.M.; Juhasz, E.; Urban, L.; Orlov, S.; Barlesi, F.; Kio, E.; et al. Randomized, Placebo-Controlled, Phase II Study of Veliparib in Combination with Carboplatin and Paclitaxel for Advanced/Metastatic Non-Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 1937–1944. [Google Scholar] [CrossRef]
- Kelly, R.J.; Rajan, A.; Force, J.; Lopez-Chavez, A.; Keen, C.; Cao, L.; Yu, Y.; Choyke, P.; Turkbey, B.; Raffeld, M.; et al. Evaluation of KRAS mutations, angiogenic biomarkers, and DCE-MRI in patients with advanced non-small-cell lung cancer receiving sorafenib. Clin. Cancer Res. 2011, 17, 1190–1199. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Johnson, M.R.; Rabe, K.G.; Brune, K.; de Andrade, M.; Goggins, M.; Rothenmund, H.; Gallinger, S.; Klein, A.; Petersen, G.M.; et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Ledermann, J.; Hall, G.; Rea, D.; Glasspool, R.; Highly, M.; Jayson, G.; Sludden, J.; Murray, J.; Jamieson, D.; et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br. J. Cancer 2016, 114, 723–730. [Google Scholar] [CrossRef]
- Murray, J.; Thomas, H.; Berry, P.; Kyle, S.; Patterson, M.; Jones, C.; Los, G.; Hostomsky, Z.; Plummer, E.R.; Boddy, A.V.; et al. Tumour cell retention of rucaparib, sustained PARP inhibition and efficacy of weekly as well as daily schedules. Br. J. Cancer 2014, 110, 1977–1984. [Google Scholar] [CrossRef]
- Drew, Y.; Mulligan, E.A.; Vong, W.; Thomas, H.D.; Kahn, S.; Kyle, S.; Mukhopadhyay, A.; Los, G.; Hostomsky, Z.; Plummer, E.R.; et al. Therapeutic Potential of Poly(ADP-ribose) Polymerase Inhibitor AG014699 in Human Cancers With Mutated or Methylated BRCA1 or BRCA2. J. Natl. Cancer Inst. 2011, 103, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef]
- A Study of Niraparib Maintenance Treatment in Patients with Advanced Ovarian Cancer Following Response on Front-Line Platinum-Based Chemotherapy. Available online: https://clinicaltrials.gov/ct2/show/NCT02655016 (accessed on 8 March 2019).
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: An updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Maintenance Rucaparib in BRCA1, BRCA2 or PALB2 Mutated Pancreatic Cancer That Has Not Progressed on Platinum-Based Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT03140670 (accessed on 8 March 2019).
- Olaparib in gBRCA Mutated Pancreatic Cancer Whose Disease Has Not Progressed on First Line Platinum-Based Chemotherapy (POLO). Available online: https://clinicaltrials.gov/ct2/show/NCT02184195 (accessed on 8 March 2019).
- A Study of Rucaparib in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT02042378 (accessed on 8 March 2019).
- Efficacy Study of Olaparib with Paclitaxel Versus Paclitaxel in Gastric Cancer Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT01063517 (accessed on 8 March 2019).
- Efficacy and Safety of Olaparib in Pretreated Patients with Measurable Colorectal Cancer, Stratified by Microsatellite Instability (MSI) Status. Available online: https://clinicaltrials.gov/ct2/show/NCT00912743 (accessed on 8 March 2019).
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- O’Donnell, R.L.; McCormick, A.; Mukhopadhyay, A.; Woodhouse, L.C.; Moat, M.; Grundy, A.; Dixon, M.; Kaufman, A.; Soohoo Elattar, A.; et al. The use of ovarian cancer cells from patients undergoing surgery to generate primary cultures capable of undergoing functional analysis. PLoS ONE 2014, 9, e90604. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J., Jr.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, R.L.; Kaufmann, A.; Woodhouse, L.; McCormick, A.; Cross, P.A.; Edmondson, R.J.; Curtin, N.J. Advanced Ovarian Cancer Displays Functional Intratumor Heterogeneity That Correlates to Ex Vivo Drug Sensitivity. Int. J. Gynecol. Cancer 2016, 26, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumours from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Bergmann, L.; Hirschfield, S.; Morris, C.; Palmeri, S.; Stone, A.; Biotherapy Development Association (BDA). Progression-free survival as an end-point in clinical trials of biotherapeutic agents. Eur. J. Cancer Suppl. 2007, 5, 23–28. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Age (years) Median (Range) | Proportion Male n (%) | Proportion Femalen (%) | Sample ID | Sampling Imepoint (Pre/post treatment) | % Pancytokeratin Positive Cells | HRR Status | Cell Growth with 10 µM Rucaparib (% Control) | Stage at Diagnosis (TNM / FIGO) | Pre-sample Treatment | Post-sample Treatment | Overall Survival (Months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COLORECTAL n = 8 (33%) | 55 (43–83) | 1 (4%) | 7 (29%) | PA001 | Pre | 100 | C | X | T4bNXM1 | Surgery | FOLFOX | 8 |
| PA002 | N/A | 95 | C | X | T4bN1M1 | Surgery | None | 2 | ||||
| PA003 | Pre | 100 | D | 23.6 | T4bN1M1 | Surgery | FOLFOX | 17 | ||||
| PA004 | Pre | 94 | C | 111.5 | T4bNXM1 | Surgery | FOLFIRI + bevacizumab | 10 | ||||
| PA005 | N/A | 100 | C | 60.3 | T4bNXM1 | None | None | 1 | ||||
| PA006 | Post | 93 | C | 39 | T4NXM1 | 1. FOLFOX + bevacizumab 2. FOLFIRI + cetuximab | Radiotherapy | 12 | ||||
| PA007 | Post | 98 | C | 20.7 | T3N2M0 | Neoadjuvant chemoradiotherapy (capecitabine); Surgery; Adjuvant CapOX; Following metastatic diagnosis: 1. Capecitabine, bevacizumab; 2. FOLFOX; 3. Irinotecan, cetuximab | 4. Phase 1 trial (gemcitabine + VX-970 ATRi) | 55 | ||||
| PA008 | Pre | 96 | D | 32.8 | T4NXM1 | Surgery | FOLFIRI + bevacizumab | 23 | ||||
| UPPER GI n = 3 (13%) | 52 (51–72) | 0 | 3 (13%) | PA009 | Post | 93 | C | 42 | T4NXM1 | Capecitabine | None | 22’ |
| PA010 | Post | 97 | D | 76.1 | T4NXM1 | EOX | None | 10 | ||||
| PA011 | Pre | 93 | C | 75.9 | T4bNXM1 | Surgery | Radiotherapy | 4 | ||||
| PANCREATIC n = 5 (21%) | 49 (48–77) | 2 (8%) | 3 (13%) | PA012 | Post | 96 | D | 49.7 | T4NXM1 | Radiotherapy | None | 11 |
| PA013 | N/A | 99 | C | 97.4 | TXNXM1 | None | None | 3 | ||||
| PA014 | N/A | 97 | D | 45.5 | T4NXM1 | None | None | 2 | ||||
| PA015 | Post | 96 | C | X | T4NXM1 | Gemcitabine | Radiotherapy | 8’ | ||||
| PA016 | Post | 99 | C | X | T4NXM1 | FOLFIRINOX | None | 4 | ||||
| HB n = 3 (13%) | 66 (44–71) | 3 (13%) | 0 | PA017 | Pre | 97 | C | 57.1 | T3NXM1 | Cisplatin/gemcitabine + panitumumab*** | None | 9 |
| PA018 | Post | 95 | C | X | T4NXM1 | Cisplatin/gemcitabine | None | 12 | ||||
| PA019 | Post | 98 | D | X | T4NXM1 | Phase 1 trial (dexanabinol, sorafenib) | None | 9 | ||||
| BREAST n = 2 (8%) | 67 (66–68) | 0 | 2 (8%) | PA020 | Post | 100 | D | 53.4 | T2N2M0 | Surgery; Adjuvant radiotherapy; Adjuvant anastrazole | 1. Letrozole 2. Capecitabine + vinorelbine 3. Exemestane + everolimus | 90 |
| PA021 | Post | 99 | C | 47.8 | UN | Paclitaxel | UN | UN | ||||
| MESOTHELIOMA n = 1 (4%) | 65 | 0 | 1 (4%) | PA022 | Post | 97 | D | 35.6 | Primary peritoneal mesothelioma (epitheloid subtype) | Carboplatin + pemetrexed | Rechallenge carboplatin + pemetrexed | 50’ |
| NEO n = 2 (8%) | 44 (26–63) | 0 | 2 (8%) | PA023 | Pre | 93 | C | 99.9 | 1C | Surgery | BEP | 58’ |
| PA024 | Pre | 98 | C | 97.4 | 3C | Surgery | Cisplatin + etoposide | 7 | ||||
| All n = 24 | 57 (26–83) | 6 (25%) | 18 (75%) | |||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentles, L.; Goranov, B.; Matheson, E.; Herriott, A.; Kaufmann, A.; Hall, S.; Mukhopadhyay, A.; Drew, Y.; Curtin, N.J.; O’Donnell, R.L. Exploring the Frequency of Homologous Recombination DNA Repair Dysfunction in Multiple Cancer Types. Cancers 2019, 11, 354. https://doi.org/10.3390/cancers11030354
Gentles L, Goranov B, Matheson E, Herriott A, Kaufmann A, Hall S, Mukhopadhyay A, Drew Y, Curtin NJ, O’Donnell RL. Exploring the Frequency of Homologous Recombination DNA Repair Dysfunction in Multiple Cancer Types. Cancers. 2019; 11(3):354. https://doi.org/10.3390/cancers11030354
Chicago/Turabian StyleGentles, Lucy, Bojidar Goranov, Elizabeth Matheson, Ashleigh Herriott, Angelika Kaufmann, Sally Hall, Asima Mukhopadhyay, Yvette Drew, Nicola J. Curtin, and Rachel L O’Donnell. 2019. "Exploring the Frequency of Homologous Recombination DNA Repair Dysfunction in Multiple Cancer Types" Cancers 11, no. 3: 354. https://doi.org/10.3390/cancers11030354
APA StyleGentles, L., Goranov, B., Matheson, E., Herriott, A., Kaufmann, A., Hall, S., Mukhopadhyay, A., Drew, Y., Curtin, N. J., & O’Donnell, R. L. (2019). Exploring the Frequency of Homologous Recombination DNA Repair Dysfunction in Multiple Cancer Types. Cancers, 11(3), 354. https://doi.org/10.3390/cancers11030354