Next Generation Sequencing in AML—On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation?

 and
and
Abstract
:1. Introduction
2. Next Generation Sequencing
2.1. Terminology
2.2. Transition of NGS from Research to Clinical Practice and Market Overview
3. NGS for the Diagnosis of AML
3.1. AML Classification and Pathogenesis
3.2. Current Guidelines Regarding NGS Analyses for the Diagnosis of AML
4. Prognostic Implication of Gene Mutations in AML
4.1. Overview of AML Prognosis and Age
4.2. Conventional Cytogenetics
4.3. Conventional Cytogenetics Refined with Analysis of Single Gene Mutations
4.4. Conventional Cytogenetics Refined with NGS Analysis of Multiple Genes
4.5. Using NGS to Predict Response to Hypomethylating Agents
4.6. Using NGS to Predict Outcome after Allogeneic Transplantation
5. Next Generation Sequencing to Guide Treatment in AML?
5.1. Overview of Current AML Treatment Strategies
5.2. Targeting FLT3 Mutations
5.2.1. Midostaurin (Novartis)
5.2.2. Gilteritinib (Astellas, Tokio, Japan)
5.2.3. Quizartinib (Daiichi Sankyo, Tokio, Japan)
5.3. Targeting IDH (Mutations)
5.3.1. Targeting IDH2 Mutations with Enasidenib (Celgene, Summit, NJ, USA)
5.3.2. Targeting IDH1 Mutations with Ivosidenib (Agios)
5.4. Targeting TP53 (Mutations) with Drugs Whose Mechanism of Action Is TP53 Independent
5.4.1. Overview of TP53 Mutations in AML
5.4.2. Targeting Bcl-2 with Venetoclax (AbbVie, Chicago, IL, USA)
5.5. Targeting JAK2 Mutations
Ruxolitinib (Novartis, Basel, Switzerland)
6. Next Generation Sequencing for Response Assessment and Disease Monitoring
6.1. Minimal Residual Disease
6.2. MRD Guided Treatment Modulation in Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Köhne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Understanding Society Scientific Group; Crawley, C.; Craig, J.; Scott, M.A.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. ICUS, IDUS, CHIP and CCUS: Diagnostic Criteria, Separation from MDS and Clinical Implications. Pathobiology 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Arock, M.; Bock, C.; George, T.I.; Galli, S.J.; Gotlib, J.; Haferlach, T.; Hoermann, G.; Hermine, O.; et al. Proposed Terminology and Classification of Pre-Malignant Neoplastic Conditions: A Consensus Proposal. EBioMedicine 2017, 26, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Patnaik, M.M.; Saeed, L.; Mudireddy, M.; Idossa, D.; Finke, C.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Targeted next-generation sequencing in myelodysplastic syndromes and prognostic interaction between mutations and IPSS-R. Am. J. Hematol. 2017, 92, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Mudireddy, M.; Lasho, T.L.; Finke, C.M.; Nicolosi, M.; Szuber, N.; Patnaik, M.M.; Pardanani, A.; Hanson, C.A.; Ketterling, R.P.; et al. Mutations and prognosis in myelodysplastic syndromes: Karyotype-adjusted analysis of targeted sequencing in 300 consecutive cases and development of a genetic risk model. Am. J. Hematol. 2018, 93, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-H.; Li, H.-Y.; Fan, S.-C.; Yuan, T.-H.; Chen, M.; Hsu, Y.-H.; Yang, Y.-H.; Li, L.-Y.; Yeh, S.-P.; Bai, L.-Y.; et al. A targeted next-generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: Implications for clinical practice. Cancer Med. 2017, 6, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, M.O.; Mirza, A.-S.; Komrokji, R.; Lancet, J.; Padron, E.; Song, J. Genetic Landscape of Acute Myeloid Leukemia Interrogated by Next-generation Sequencing: A Large Cancer Center Experience. Cancer Genomics Proteomics 2018, 15, 121–126. [Google Scholar] [PubMed]
- Ruffalo, M.; Husseinzadeh, H.; Makishima, H.; Przychodzen, B.; Ashkar, M.; Koyutürk, M.; Maciejewski, J.P.; LaFramboise, T. Whole-exome sequencing enhances prognostic classification of myeloid malignancies. J. Biomed. Inform. 2015, 58, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Au, C.H.; Wa, A.; Ho, D.N.; Chan, T.L.; Ma, E.S.K. Clinical evaluation of panel testing by next-generation sequencing (NGS) for gene mutations in myeloid neoplasms. Diagn. Pathol. 2016, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Hou, G.; Wang, L.; Lv, N.; Xu, Y.; Xu, Y.; Wang, X.; Xuan, Z.; Jing, Y.; et al. Mutational spectrum and risk stratification of intermediate-risk acute myeloid leukemia patients based on next-generation sequencing. Oncotarget 2016, 7, 32065–32078. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Landgren, O. MGUS and Smoldering Multiple Myeloma: Diagnosis and Epidemiology. Cancer Res. Ther. 2016, 169, 3–12. [Google Scholar]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Walker, H.; Harrison, G.; Oliver, F.; Chatters, S.; Harrison, C.J.; Wheatley, K.; Burnett, A.K.; Goldstone, A.H.; Medical Research Council Adult Leukemia Working Party. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): Analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001, 98, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Ruppert, A.S.; Mrózek, K.; Mayer, R.J.; Stone, R.M.; Carroll, A.J.; Powell, B.L.; Moore, J.O.; Pettenati, M.J.; Koduru, P.R.K.; et al. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: A cancer and leukemia group B study. J. Clin. Oncol. 2005, 23, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Herold, T.; Rothenberg-thurley, M.; Amler, S.; Sauerland, M.C.; Dennis, G.; Ksienzyk, B.; Zellmeier, E.; Hartmann, L.; Greif, P.A.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2017, 128, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, C.D.; Lawrence, D.; Byrd, J.C.; Carroll, A.; Pettenati, M.J.; Tantravahi, R.; Patil, S.R.; Davey, F.R.; Berg, D.T.; Schiffer, C.A.; et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998, 58, 4173–4179. [Google Scholar] [PubMed]
- Byrd, J.C.; Mrózek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Slovak, M.L.; Kopecky, K.J.; Cassileth, P.A.; Harrington, D.H.; Theil, K.S.; Mohamed, A.; Paietta, E.; Willman, C.L.; Head, D.R.; Rowe, J.M.; et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: A Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 2000, 96, 4075–4083. [Google Scholar] [PubMed]
- Delaunay, J.; Vey, N.; Leblanc, T.; Fenaux, P.; Rigal-Huguet, F.; Witz, F.; Lamy, T.; Auvrignon, A.; Blaise, D.; Pigneux, A.; et al. Prognosis of inv(16)/t(16;16) acute myeloid leukemia (AML): A survey of 110 cases from the French AML Intergroup. Blood 2003, 102, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Benner, A.; Krauter, J.; Büchner, T.; Sauerland, C.; Ehninger, G.; Schaich, M.; Mohr, B.; Niederwieser, D.; Krahl, R.; et al. Individual patient data-based meta-analysis of patients aged 16 to 60 years with core binding factor acute myeloid leukemia: A survey of the German Acute Myeloid Leukemia Intergroup. J. Clin. Oncol. 2004, 22, 3741–3750. [Google Scholar] [CrossRef] [PubMed]
- Keating, M.J.; Smith, T.L.; Kantarjian, H.; Cork, A.; Walters, R.; Trujillo, J.M.; McCredie, K.B.; Gehan, E.A.; Freireich, E.J. Cytogenetic pattern in acute myelogenous leukemia: A major reproducible determinant of outcome. Leukemia 1988, 2, 403–412. [Google Scholar] [PubMed]
- Ramos, F.; Thépot, S.; Pleyer, L.; Maurillo, L.; Itzykson, R.; Bargay, J.; Stauder, R.; Venditti, A.; Seegers, V.; Martínez-Robles, V.; et al. Azacitidine frontline therapy for unfit acute myeloid leukemia patients: Clinical use and outcome prediction. Leuk. Res. 2015, 39, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and therapeutic implications. Hematology 2016, 2016, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Pastore, F.; Dufour, A.; Benthaus, T.; Metzeler, K.H.; Maharry, K.S.; Schneider, S.; Ksienzyk, B.; Mellert, G.; Zellmeier, E.; Kakadia, P.M.; et al. Combined molecular and clinical prognostic index for relapse and survival in cytogenetically normal acute myeloid leukemia. J. Clin. Oncol. 2014, 32, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Marceau-Renaut, A.; Boissel, N.; Petit, A.; Bucci, M.; Geffroy, S.; Lapillonne, H.; Renneville, A.; Ragu, C.; Figeac, M.; et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 2016, 127, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Papaemmanuil, E.; Martincorena, I.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Heuser, M.; Thol, F.; Bolli, N.; Ganly, P.; et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat. Genet. 2017, 49, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Huet, S.; Paubelle, E.; Lours, C.; Grange, B.; Courtois, L.; Chabane, K.; Charlot, C.; Mosnier, I.; Simonet, T.; Hayette, S.; et al. Validation of the prognostic value of the knowledge bank approach to determine AML prognosis in real life. Blood 2018, 132, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Perez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Itzykson, R.; Renneville, A.; de Renzis, B.; Dreyfus, F.; Laribi, K.; Bouabdallah, K.; Vey, N.; Toma, A.; Recher, C.; et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: A phase 2 trial. Blood 2011, 118, 3824–3831. [Google Scholar] [CrossRef] [PubMed]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Kim, Y.-J.; Yim, S.-H.; Kim, H.-J.; Kwon, Y.-R.; Hur, E.-H.; Goo, B.-K.; Choi, Y.-S.; Lee, S.H.; Chung, Y.-J.; et al. Somatic mutations predict outcomes of hypomethylating therapy in patients with myelodysplastic syndrome. Oncotarget 2016, 7, 55264–55275. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 2012, 26, 1106–1107. [Google Scholar] [CrossRef] [PubMed]
- Craddock, C.F.; Houlton, A.E.; Quek, L.S.; Ferguson, P.; Gbandi, E.; Roberts, C.; Metzner, M.; Garcia-Martin, N.; Kennedy, A.; Hamblin, A.; et al. Outcome of azacitidine therapy in acute myeloid leukemia is not improved by concurrent vorinostat therapy but is predicted by a diagnostic molecular signature. Clin. Cancer Res. 2017, 23, 6430–6440. [Google Scholar] [CrossRef] [PubMed]
- Kuendgen, A.; Müller-Thomas, C.; Lauseker, M.; Haferlach, T.; Urbaniak, P.; Schroeder, T.; Brings, C.; Wulfert, M.; Meggendorfer, M.; Hildebrandt, B.; et al. Efficacy of azacitidine is independent of molecular and clinical characteristics—An analysis of 128 patients with myelodysplastic syndromes or acute myeloid leukemia and a review of the literature. Oncotarget 2018, 9, 27882–27894. [Google Scholar] [CrossRef] [PubMed]
- Luskin, M.R.; Carroll, M.; Lieberman, D.; Morrissette, J.J.D.; Zhao, J.; Crisalli, L.; Roth, D.B.; Luger, S.M.; Porter, D.L.; Reshef, R. Clinical Utility of Next-Generation Sequencing for Oncogenic Mutations in Patients with Acute Myeloid Leukemia Undergoing Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Middeke, J.M.; Herold, S.; Rücker-Braun, E.; Berdel, W.E.; Stelljes, M.; Kaufmann, M.; Schäfer-Eckart, K.; Baldus, C.D.; Stuhlmann, R.; Ho, A.D.; et al. TP53 mutation in patients with high-risk acute myeloid leukaemia treated with allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2016, 172, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.; Ferguson, P.; Metzner, M.; Ahmed, I.; Kennedy, A.; Garnett, C.; Jeffries, S.; Walter, C.; Piechocki, K.; Timbs, A.; et al. Mutational analysis of disease relapse in patients allografted for acute myeloid leukemia. Blood 2016, 1, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine Versus Patient Choice, With Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients With Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Schuh, A.C.; Döhner, H.; Pleyer, L.; Seymour, J.F.; Fenaux, P.; Dombret, H. Azacitidine in adult patients with acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2017, 116, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Stauder, R.; Burgstaller, S.; Schreder, M.; Tinchon, C.; Pfeilstocker, M.; Steinkirchner, S.; Melchardt, T.; Mitrovic, M.; Girschikofsky, M.; et al. Azacitidine in patients with WHO-defined AML—Results of 155 patients from the Austrian Azacitidine Registry of the AGMT-Study Group. J. Hematol. Oncol. 2013, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Burgstaller, S.; Girschikofsky, M.; Linkesch, W.; Stauder, R.; Pfeilstocker, M.; Schreder, M.; Tinchon, C.; Sliwa, T.; Lang, A.; et al. Azacitidine in 302 patients with WHO-defined acute myeloid leukemia: Results from the Austrian Azacitidine Registry of the AGMT-Study Group. Ann. Hematol. 2014, 93, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; Sill, H.; Schlick, K.; Thaler, J.; Halter, B.; Machherndl-Spandl, S.; Zebisch, A.; et al. Azacitidine front-line in 339 patients with myelodysplastic syndromes and acute myeloid leukaemia: Comparison of French-American-British and World Health Organization classifications. J. Hematol. Oncol. 2016, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Döhner, H.; Dombret, H.; Seymour, J.; Schuh, A.; Beach, C.; Swern, A.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; et al. Azacitidine for Front-Line Therapy of Patients with AML: Reproducible Efficacy Established by Direct Comparison of International Phase 3 Trial Data with Registry Data from the Austrian Azacitidine Registry of the AGMT Study Group. Int. J. Mol. Sci. 2017, 18, 415. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Stone, R.M.; DeAngelo, D.J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E.J.; Schiller, G.J.; et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010, 28, 4339–4345. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Perl, A.E.; Döhner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.-P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J. Efficacy and Safety of Single-Agent Quizartinib (Q), a Potent and Selective FLT3 Inhibitor (FLT3i), in Patients (pts) with FLT3-Internal Tandem Duplication (FLT3-ITD)-Mutated Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) Enrolled in the Global, Phase 3, Randomized Controlled Quantum-R Trial. Blood 2018, 132. [Google Scholar]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J. Clin. Oncol. 2013, 31, 3681–3687. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Knapper, S.; Russell, N.; Gilkes, A.; Hills, R.K.; Gale, R.E.; Cavenagh, J.D.; Jones, G.; Kjeldsen, L.; Grunwald, M.R.; Thomas, I.; et al. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood 2017, 129, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, W.; Kayser, S.; Kebenko, M.; Janning, M.; Krauter, J.; Schittenhelm, M.; Götze, K.; Weber, D.; Göhring, G.; Teleanu, V.; et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br. J. Haematol. 2015, 169, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, J.K.; Kantarjian, H.M.; Borthakur, G.; Thompson, P.A.; Konopleva, M.; Daver, N.; Pemmaraju, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.; et al. Results of a Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients (Pts) with Activating FLT3 Mutations. Blood 2014, 124, 389. [Google Scholar]
- Ohanian, M.; Kantarjian, H.M.; Borthakur, G.; Kadia, T.M.; Konopleva, M.; Garcia-Manero, G.; Estrov, Z.; Ferrajoli, A.; Takahashi, K.; Jabbour, E.J.; et al. Efficacy of a Type I FLT3 Inhibitor, Crenolanib, with Idarubicin and High-Dose Ara-C in Multiply Relapsed/Refractory FLT3+ AML. Blood 2016, 128, 2744. [Google Scholar]
- Iyer, S.P.; Jethava, Y.; Karanes, C.; Eckardt, J.R.; Collins, R. Safety Study of Salvage Chemotherapy High-Dose Ara-C/Mitoxantrone (HAM) and Type I FLT3-TKI Crenolanib in First Relapsed/Primary Refractory AML. Blood 2016, 128, 3983. [Google Scholar]
- Wang, E.S.; Tallman, M.S.; Stone, R.M.; Walter, R.B.; Karanes, C.; Jain, V.; Collins, R.H. Low Relapse Rate in Younger Patients ≤ 60 Years Old with Newly Diagnosed FLT3-Mutated Acute Myeloid Leukemia (AML) Treated with Crenolanib and Cytarabine/Anthracycline Chemotherapy. Blood 2017, 130, 566. [Google Scholar]
- Wang, E.S.; Stone, R.M.; Tallman, M.S.; Walter, R.B.; Eckardt, J.R.; Collins, R. Crenolanib, a Type I FLT3 TKI, Can be Safely Combined with Cytarabine and Anthracycline Induction Chemotherapy and Results in High Response Rates in Patients with Newly Diagnosed FLT3 Mutant Acute Myeloid Leukemia (AML). Blood 2016, 128, 1071. [Google Scholar]
- Ravandi, F.; Cortes, J.E.; Jones, D.; Faderl, S.; Garcia-Manero, G.; Konopleva, M.Y.; O’Brien, S.; Estrov, Z.; Borthakur, G.; Thomas, D.; et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Serve, H.; Krug, U.; Wagner, R.; Sauerland, M.C.; Heinecke, A.; Brunnberg, U.; Schaich, M.; Ottmann, O.; Duyster, J.; Wandt, H.; et al. Sorafenib in Combination With Intensive Chemotherapy in Elderly Patients With Acute Myeloid Leukemia: Results from a Randomized, Placebo-Controlled Trial. J. Clin. Oncol. 2013, 31, 3110–3118. [Google Scholar] [CrossRef] [PubMed]
- Rautenberg, C.; Nachtkamp, K.; Dienst, A.; Schmidt, P.V.; Heyn, C.; Kondakci, M.; Germing, U.; Haas, R.; Kobbe, G.; Schroeder, T. Sorafenib and azacitidine as salvage therapy for relapse of FLT3-ITD mutated AML after allo-SCT. Eur. J. Haematol. 2017, 98, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Röllig, C.; Serve, H.; Hüttmann, A.; Noppeney, R.; Müller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.; Kunzmann, H.V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A. Sorafenib as Maintenance Therapy Post Allogeneic Stem Cell Transplantation for FLT3-ITD Positive AML: Results from the Randomized, Double-Blind, Placebo-Controlled Multicentre Sormain Trial. In Proceedings of the ASH Annual Meeting, Washington, DC, USA, 3 December 2018. [Google Scholar]
- Eytan, M.S.; Courtney, D.D.; Daniel, A.P.; Amir, T.F.; Gail, J.R.; Jessica, K.A.; Richard, M.S.; Daniel, J.D.; Ross, L.L.; Ian, W.F.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2018, 130, 722–732. [Google Scholar]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; Raza, A.; Carter, T.H.; Claxton, D.; Erba, H.; DeAngelo, D.J.; Tallman, M.S.; Goard, C.; Borthakur, G. A multicenter phase l/ll study of obatoclax mesylate administered as a 3- Or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS ONE 2014, 9, e108694. [Google Scholar] [CrossRef] [PubMed]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Löffler, H.; Sauerland, C.M.; Serve, H.; Büchner, T.; et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002, 100, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2012, 99, 4326–4335. [Google Scholar] [CrossRef]
- Whitman, S.P.; Archer, K.J.; Feng, L.; Baldus, C.; Becknell, B.; Carlson, B.D.; Carroll, A.J.; Mrózek, K.; Vardiman, J.W.; George, S.L.; et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: A cancer and leukemia group B study. Cancer Res. 2001, 61, 7233–7239. [Google Scholar] [PubMed]
- Weisberg, E.; Barrett, R.; Liu, Q.; Stone, R.; Gray, N.; Griffin, J.D. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist. Updat. 2009, 12, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Zuffa, E.; Franchini, E.; Papayannidis, C.; Baldazzi, C.; Simonetti, G.; Testoni, N.; Abbenante, C.M.; Paolini, S.; Sartor, C.; Parisi, S.; et al. Revealing very small FLT3 ITD mutated clones by ultra-deep sequencing analysis has important clinical implications in AML patients. Oncotarget 2015, 6, 31284–31294. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-K.; Mishra, A.; Chandler, J.; Whitman, S.P.; Marcucci, G.; Caligiuri, M.A. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: Implications for Axl as a potential therapeutic target. Blood 2013, 121, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves Gilteritinib for Relapsed or Refractory Acute Myeloid Leukemia (AML) with a FLT3 Mutation. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm627045.htm (accessed on 15 February 2019).
- Nassereddine, S.; Lap, C.J.; Haroun, F.; Tabbara, I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann. Hematol. 2017, 96, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Ratcliffe, P.J. CANCER: Puzzling Patterns of Predisposition. Science 2009, 324, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Paschka, P.; Späth, D.; Habdank, M.; Köhne, C.-H.; Germing, U.; von Lilienfeld-Toal, M.; Held, G.; Horst, H.-A.; Haase, D.; et al. TET2 Mutations in Acute Myeloid Leukemia (AML): Results From a Comprehensive Genetic and Clinical Analysis of the AML Study Group. J. Clin. Oncol. 2012, 30, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; AG221-C-001 Study Investigators. Differentiation Syndrome Associated With Enasidenib, a Selective Inhibitor of Mutant Isocitrate Dehydrogenase 2: Analysis of a Phase 1/2 Study. JAMA Oncol. 2018, 4, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Platt, M.Y.; Fathi, A.T.; Borger, D.R.; Brunner, A.M.; Hasserjian, R.P.; Balaj, L.; Lum, A.; Yip, S.; Dias-Santagata, D.; Zheng, Z.; et al. Detection of Dual IDH1 and IDH2 Mutations by Targeted Next-Generation Sequencing in Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Mol. Diagn. 2015, 17, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; McGraw, K.L.; Sallman, D.A.; List, A.F. The role of p53 in myelodysplastic syndromes and acute myeloid leukemia: Molecular aspects and clinical implications. Leuk. Lymphoma 2017, 58, 1777–1790. [Google Scholar] [CrossRef] [PubMed]
- Yanada, M.; Yamamoto, Y.; Iba, S.; Okamoto, A.; Inaguma, Y.; Tokuda, M.; Morishima, S.; Kanie, T.; Mizuta, S.; Akatsuka, Y.; et al. TP53 mutations in older adults with acute myeloid leukemia. Int. J. Hematol. 2016, 103, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S. Patterns of mutations in TP53 mutated AML. Best Pract. Res. Clin. Haematol. 2018, 31, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.S.; Cohen, G.M. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br. J. Haematol. 2013, 163, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Pollyea, D.A. Shutting Down Acute Myeloid Leukemia and Myelodysplastic Syndrome with BCL-2 Family Protein Inhibition. Curr. Hematol. Malig. Rep. 2018, 13, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.-M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Kontro, M.; Kumar, A.; Majumder, M.M.; Eldfors, S.; Parsons, A.; Pemovska, T.; Saarela, J.; Yadav, B.; Malani, D.; Fløisand, Y.; et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia 2017, 31, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Ritchie, E.K.; Sayar, H.; Lancet, J.E.; Craig, M.D.; Vey, N.; Strickland, S.A.; Schiller, G.J.; Jabbour, E.; Erba, H.P.; et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): A randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol. 2015, 16, 1025–1036. [Google Scholar] [CrossRef]
- Nijenhuis, C.M.; Lucas, L.; Rosing, H.; Huitema, A.D.R.; Mergui-Roelvink, M.; Jamieson, G.C.; Fox, J.A.; Mould, D.R.; Schellens, J.H.M.; Beijnen, J.H. Metabolism and disposition of the anticancer quinolone derivative vosaroxin, a novel inhibitor of topoisomerase II. Invest. New Drugs 2017, 35, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Kantarjian, H.; Garcia-Manero, G.; Jabbour, E.; Borthakur, G.; Brandt, M.; Pierce, S.; Vaughan, K.; Ning, J.; Nogueras González, G.M.; et al. Vosaroxin in combination with decitabine in newly diagnosed older patients with acute myeloid leukemia or high-risk myelodysplastic syndrome. Haematologica 2017, 102, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Stuart, R.K.; Cripe, L.D.; Maris, M.B.; Cooper, M.A.; Stone, R.M.; Dakhil, S.R.; Turturro, F.; Stock, W.; Mason, J.; Shami, P.J.; et al. REVEAL-1, a phase 2 dose regimen optimization study of vosaroxin in older poor-risk patients with previously untreated acute myeloid leukaemia. Br. J. Haematol. 2015, 168, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Griesshammer, M.; Saydam, G.; Palandri, F.; Benevolo, G.; Egyed, M.; Callum, J.; Devos, T.; Sivgin, S.; Guglielmelli, P.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythemia vera without splenomegaly: 80-week follow-up from the RESPONSE-2 trial. Ann. Hematol. 2018, 97, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Kantarjian, H.; Kadia, T.; Cortes, J.; Borthakur, G.; Newberry, K.; Garcia-Manero, G.; Ravandi, F.; Jabbour, E.; Dellasala, S.; et al. A Phase I/II Study of the Janus Kinase (JAK)1 and 2 Inhibitor Ruxolitinib in Patients With Relapsed or Refractory Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Eghtedar, A.; Verstovsek, S.; Estrov, Z.; Burger, J.; Cortes, J.; Bivins, C.; Faderl, S.; Ferrajoli, A.; Borthakur, G.; George, S.; et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood 2012, 119, 4614–4618. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Kantarjian, H.M.; Huang, X.; Pierce, S.A.; Sun, Z.; Gundacker, H.M.; Ravandi, F.; Faderl, S.H.; Tallman, M.S.; Appelbaum, F.R.; et al. Effect of Complete Remission and Responses Less Than Complete Remission on Survival in Acute Myeloid Leukemia: A Combined Eastern Cooperative Oncology Group, Southwest Oncology Group, and M. D. Anderson Cancer Center Study. J. Clin. Oncol. 2010, 28, 1766–1771. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, C.D.; Estey, E.; Pleyer, L.; Schuh, A.C.; Stein, E.M.; Tallman, M.S.; Wei, A. Time to repeal and replace response criteria for acute myeloid leukemia? Blood Rev. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Freeman, S.D. Defining minimal residual disease in acute myeloid leukemia: Which platforms are ready for “prime time”? Blood 2014, 124, 3345–3355. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.J.; Schuurhuis, G.J. MRD in AML: It is time to change the definition of remission. Best Pract. Res. Clin. Haematol. 2014, 27, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Terwijn, M.; van Putten, W.L.J.; Kelder, A.; van der Velden, V.H.J.; Brooimans, R.A.; Pabst, T.; Maertens, J.; Boeckx, N.; de Greef, G.E.; Valk, P.J.M.; et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: Data from the HOVON/SAKK AML 42A study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Schlenk, R.F.; Jensen, K.-O.; Tschürtz, F.; Corbacioglu, A.; Gaidzik, V.I.; Paschka, P.; Onken, S.; Eiwen, K.; Habdank, M.; et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: A study from the German-Austrian acute myeloid leukemia study group. J. Clin. Oncol. 2011, 29, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E.; et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 2013, 121, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, A.; Nadarajah, N.; Alpermann, T.; Grossmann, V.; Schindela, S.; Dicker, F.; Roller, A.; Kern, W.; Haferlach, C.; Schnittger, S.; et al. Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia 2014, 28, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Debarri, H.; Lebon, D.; Roumier, C.; Cheok, M.; Marceau-Renaut, A.; Nibourel, O.; Geffroy, S.; Helevaut, N.; Rousselot, P.; Gruson, B.; et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: A study by the Acute Leukemia French Association. Oncotarget 2015, 6, 42345–42353. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.-H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S.; et al. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3-ITD or NPM1 mutations. Genes. Chromosom. Cancer 2012, 51, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA 2015, 314, 811. [Google Scholar] [CrossRef] [PubMed]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-H.; Zhang, X.-H.; Qin, Y.-Z.; Liu, D.-H.; Jiang, H.; Chen, H.; Jiang, Q.; Xu, L.-P.; Lu, J.; Han, W.; et al. MRD-directed risk stratification treatment may improve outcomes of t(8;21) AML in the first complete remission: Results from the AML05 multicenter trial. Blood 2013, 121, 4056–4062. [Google Scholar] [CrossRef] [PubMed]
- Balsat, M.; Renneville, A.; Thomas, X.; de Botton, S.; Caillot, D.; Marceau, A.; Lemasle, E.; Marolleau, J.-P.; Nibourel, O.; Berthon, C.; et al. Postinduction Minimal Residual Disease Predicts Outcome and Benefit From Allogeneic Stem Cell Transplantation in Acute Myeloid Leukemia With NPM1 Mutation: A Study by the Acute Leukemia French Association Group. J. Clin. Oncol. 2017, 35, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Middeke, J.M.; Sockel, K.; Herbst, R.; Wolf, D.; Baldus, C.D.; Oelschlägel, U.; Mütherig, A.; Fransecky, L.; Noppeney, R.; et al. Measurable residual disease-guided treatment with azacitidine to prevent haematological relapse in patients with myelodysplastic syndrome and acute myeloid leukaemia (RELAZA2): An open-label, multicentre, phase 2 trial. Lancet Oncol. 2018, 19, 1668–1679. [Google Scholar] [CrossRef]
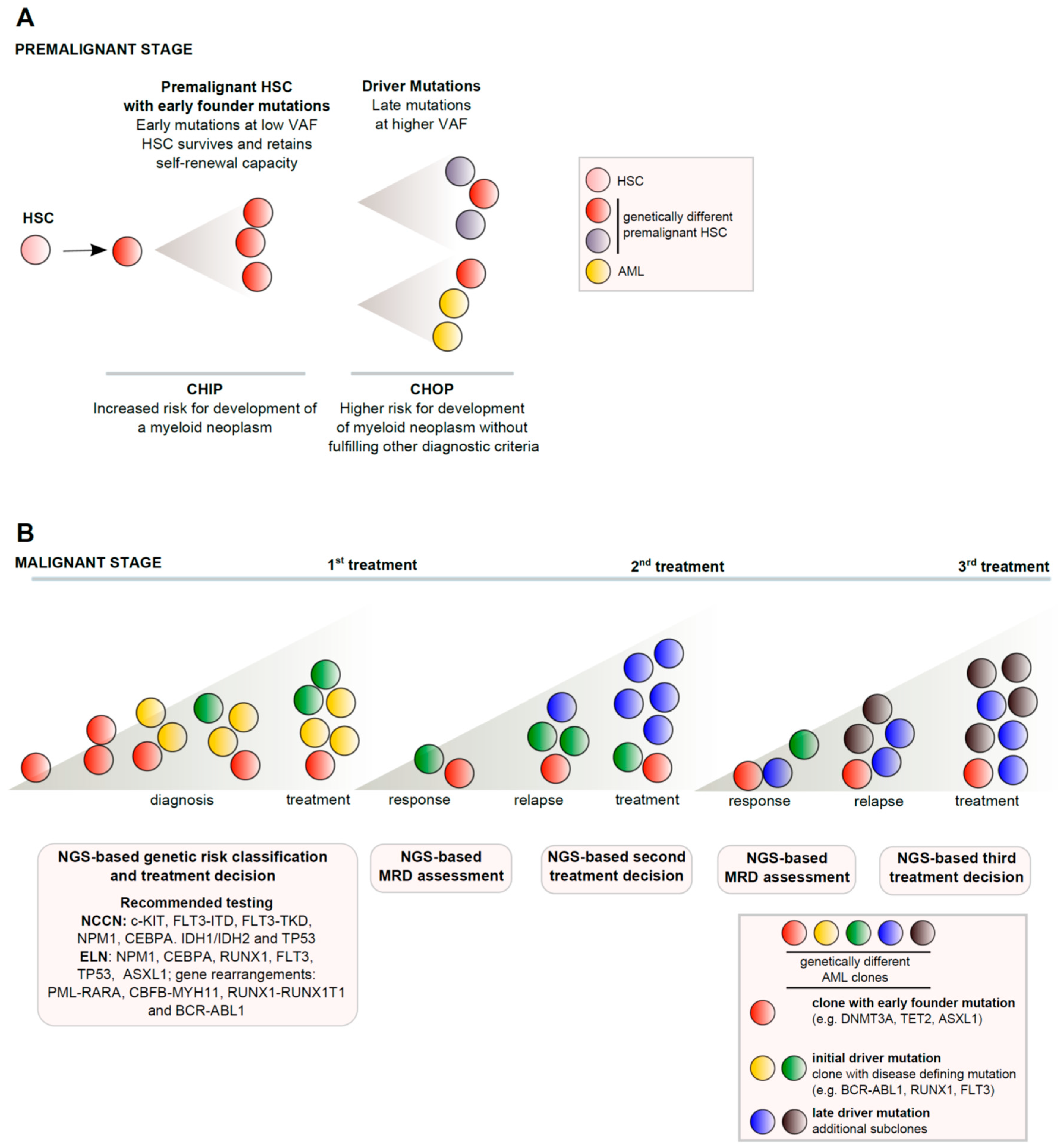
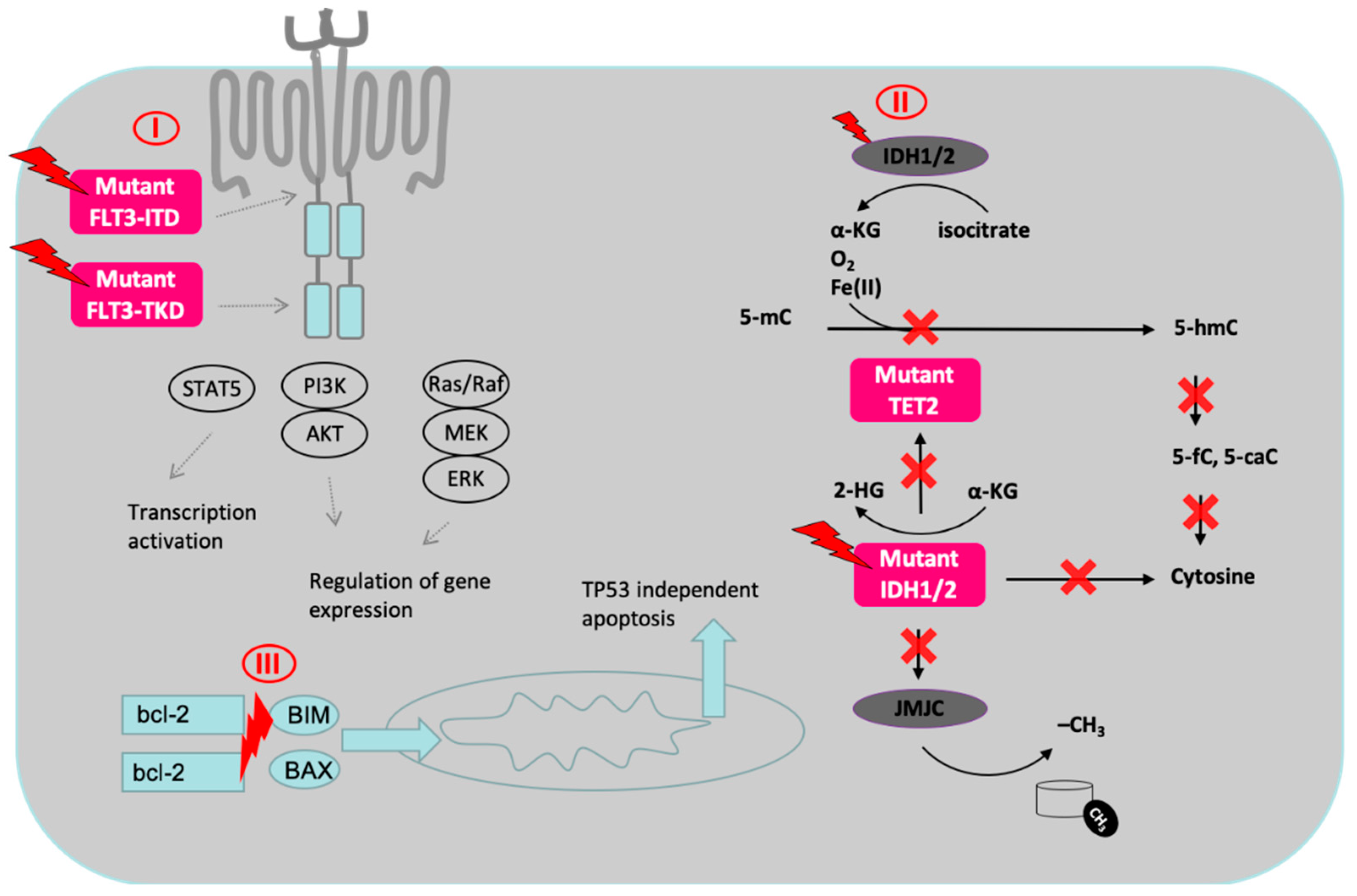

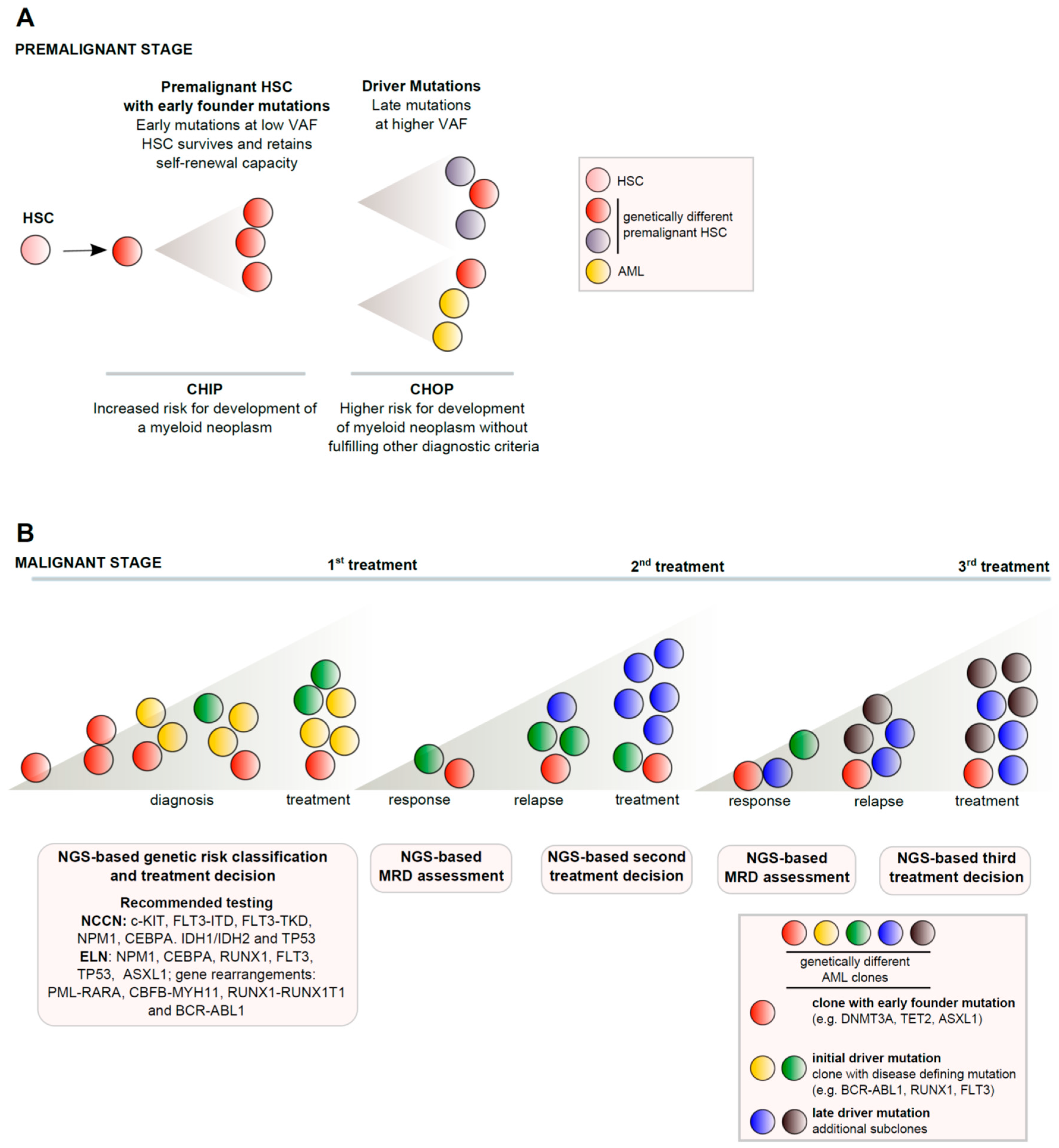{kind=link}
{kind=link}
| Qiagen Human Myeloid Neoplasms Panel 1 | Illumina AmpliSeq Myeloid Panel 2 | Quest Diagnostics LeukoVantage Panel 3 | Oxford Gene Technology SureSeq myPanel NGS Custom AML 1 |
|---|---|---|---|
| ASXL1 (full) | ASXL1 (full) | ASXL1 | ASXL1 (full) |
| CEBPA (full) | CEBPA (full) | CEBPA | CEBPA (full) |
| DNMT3A (full) | DNMT3A (hotspot) | DNMT3A | DNMT3A (full) |
| FLT3 (full) | FLT3 (hotspot) | FLT3 | FLT3 (full) |
| IDH1 (full) | IDH1 (hotspot) | IDH1 | IDH1 (full) |
| IDH2 (full) | IDH2 (hotspot) | IDH2 | IDH2 (full) |
| KIT (full) | KIT (hotspot) | KIT | KIT (full) |
| KMT2A (full) | KMT2A (fusion) | KMT2A | KMT2A (full) |
| KRAS (full) | KRAS (hotspot) | KRAS | KRAS (full) |
| NPM1 (full) | NPM1 (hotspot) | NPM1 | NPM1 (full) |
| NRAS (full) | NRAS (hotspot) | NRAS | NRAS (full) |
| RUNX1 (full) | RUNX1 (full) | RUNX1 | RUNX1 (full) |
| TET2 (full) | TET2 (full) | TET2 | TET2 (full) |
| TP53 (full) | TP53 (full) | TP53 | TP53 (full) |
| U2AF1 (full) | U2AF1 (hotspot) | U2AF1 | U2AF1 (full) |
| WT1 (full) | WT1 (hotspot) | WT1 | WT1 (full) |
| BCOR (full) | BCOR (full) | - | BCOR (full) |
| CALR (full) | CALR (full) | CALR | - |
| CBL (full) | CBL (hotspot) | CBL | - |
| CSF3R (full) | CSF3R (hotspot) | CSF3R | - |
| ETV6 (full) | ETV6 (full) | - | ETV6 (full) |
| EZH2 (full) | EZH2 (full) | EZH2 | - |
| GATA1 (full) | - | GATA1 | GATA1 (full) |
| JAK2 (full) | JAK2 (hotspot) | JAK2 | - |
| MPL (full) | MPL (hotspot) | MPL | - |
| PHF6 (full) | PHF6 (full) | - | PHF6 (full) |
| PTPN11 (full) | PTPN11 (hotspot) | PTPN11 | - |
| SETBP1 (full) | SETBP1 (hotspot) | SETBP1 | - |
| SF3B1 (full) | SF3B1 (hotspot) | SF3B1 | - |
| SRSF2 (full) | SRSF2 (hotspot) | SRSF2 | - |
| ZRSR2 (full) | ZRSR2 (full) | ZRSR2 | - |
| ABL1 (full) | ABL1 (hotspot) | - | - |
| BRAF (full) | BRAF (hotspot) | - | - |
| CREBBP (full) | CREBBP (fusion) | - | - |
| DDX41 (full) | - | DDX41 | - |
| EGFR (full) | EGFR (fusion) | - | - |
| GATA2 (full) | GATA2 (hotspot) | - | - |
| HRAS (full) | HRAS (hotspot) | - | - |
| IKZF1 (full) | IKZF1(full) | - | - |
| KMD6A (full) | - | KMD6A | - |
| MYC (full) | MYC (expression) | - | - |
| MYD88 (full) | MYD88 (hotspot) | - | - |
| NF1 (full) | NF1 (full) | - | - |
| NTRK3 (full) | NTRK3 (fusion) | - | - |
| PDGFRA (full) | PDGFRA (fusion) | - | - |
| PRPF8 (full) | PRPF8 (full) | - | - |
| RB1 (full) | RB1 (full) | - | - |
| SH2B3 (full) | SH2B3 (full) | - | - |
| SMC1A (full) | SMC1A (expression) | - | - |
| STAG2 (full) | STAG2 (full) | - | - |
| 91 genes available |
| Author | Papaemanuil [1] | Tefferi [14] | Gangat [15] | Han Lin [16] | Hussaini [17] | Ruffalo [18] | Ley [2] | Lindsley [19] | Lindsley [19] | Lindsley [19] | Chun Ha [20] | Wang [21] | Welch [22] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Year Published | 2016 | 2017 | 2018 | 2016 | 2018 | 2015 | 2013 | 2014 | 2014 | 2014 | 2016 | 2016 | 2016 |
| n pts | 1540 | 179 | 300 | 112 | 187 | 274 | 200 | 93 1 | no data 2 | 101 3 | 60 | 95 | 54 |
| Median Age | 18–65 | 73 | <70 | 43 | no data | 61,9 | 55 | 62 | 62 | 62 | 50 | 45 | 74 |
| n-Genes in Panel | 111 | 27 | 27 | 260 | 21 | 71 | WGS/WES | 82 | 82 | 82 | 54 | 410 | 264 |
| Gene | %pts | %pts | %pts | %pts | %pts | %pts | %pts | %pts | %pts | %pts | %pts | %pts | %pts |
| TET2 | 13.3 | 25 | 26 | 10 | 15.3 | 14.0 | 8.0 | 20.0 | 9.0 | 14.0 | 8.0 | 9.5 | 14.8 |
| ASXL1 | 4.61 | 30 | 27 | 16 | 20.7 | 5.0 | 2.5 | 32.0 | 3.0 | no data | 8.0 | 9.5 | 11.1 |
| SMC1A | - | - | - | no data | - | - | no data | 3.0 | 4.0 | 3.0 | no data | no data | 3.7 |
| BCOR | 2.34 | - | - | no data | - | 2.0 | no data | 8.0 | 2.0 | 1.0 | no data | - | 5.6 |
| DNMT3A | 24.9 | 10 | 13 | 15 | 14.8 | 21.0 | 26.0 | 19.0 | 28.0 | 27.0 | 8.0 | 16.8 | 14.8 |
| IDH2 | 9.9 | 6 | 4 | 12 | 12 | 8.0 | 10.0 | 11.0 | 11.0 | 17.0 | 14.0 | 11.6 | 16.7 |
| TP53-Others | 7.21 | 13 | 12 | no data | 14.4 | 8.0 | 8.0 | 15.0 | 9.0 | 23.0 | 8.0 | 5.3 | 25.9 |
| EZH2 | 3.12 | 4 | 3 | no data | - | 3.0 | 1.5 | 9.0 | 2.0 | 3.0 | 2.0 | no data | 1.9 |
| KAT6A | - | - | - | no data | - | - | no data | - | - | - | - | - | 0.0 |
| IDH1 | 6.88 | 3 | 3 | no data | 10 | 8.0 | 9.5 | 11.0 | 11.0 | 17.0 | 6.0 | 4.2 | 9.3 |
| JAK3 | no data | - | - | no data | - | - | no data | - | - | - | no data | no data | 0.0 |
| KIT | 4.61 | 2 | no data | no data | 10 | 2.0 | 4.0 | 3.0 | no data | 2.0 | 4.0 | 2.1 | 5.6 |
| RUNX1 | 9.8 | 11 | 10 | no data | 15.2 | 9.0 | 10.0 | 31.0 | 11.0 | 11.0 | - | 5.3 | 16.7 |
| SRSF2 | 6.04 | 16 | 13 | no data | - | 8.0 | no data | 20.0 | 1.0 | 10.0 | no data | 4.2 | 18.5 |
| NF1 | 2.53 | - | - | no data | - | 3.0 | no data | 6.0 | 4.0 | 4.0 | - | 1.1 | 1.9 |
| BCORL1 | - | - | - | no data | - | - | no data | no data | no data | no data | no data | - | 0.0 |
| WT1 | 5.26 | - | - | 11 | - | 4.0 | 6.0 | no data | no data | 3.0 | no data | 11.6 | 7.4 |
| FLT3 others | 37.4 | 0.5 | no data | 21 | 11 | 16.0 | 28.0 | 19.0 | 28.0 | 16.0 | 32.0 | 18.9 | 5.6 |
| NPM1 | 28.6 | no data | no data | no data | 11 | 16.0 | 27.0 | 5.0 | 30.0 | 16.0 | 24.0 | 21.5 | 11.1 |
| IKZF1 | no data | no data | no data | no data | - | - | no data | no data | no data | no data | 2.0 | 1.1 | 0.0 |
| KRAS | 5.19 | - | - | no data | - | 4.0 | 12.0 | 8.0 | 4.0 | 11.0 | no data | 3.2 | 3.7 |
| NRAS | 19.0 | no data | no data | no data | 11.9 | 6.0 | 12.0 | 23.0 | 8.0 | 13.0 | 2.0 | 12.6 | 9.3 |
| ATRX | 0.39 | - | - | no data | - | - | no data | no data | no data | no data | - | - | 0.0 |
| ZRSR2 | 0.78 | no data | no data | no data | 10 | - | no data | 8.0 | no data | 1.0 | 2.0 | - | 0.0 |
| SF3B1 | 2.60 | 20 | 30 | no data | 10 | 5.0 | no data | 11.0 | 1.0 | 3.0 | 2.0 | 1.1 | 7.4 |
| STAG2 | 4.48 | - | - | no data | - | 4.0 | no data | 14.0 | 2.0 | 6.0 | 4.0 | 5.3 | 5.6 |
| U2AF1 | 2.47 | 16 | no data | no data | 10 | 6.0 | no data | 16.0 | 4.0 | 5.0 | no data | 7.4 | 9.3 |
| SETBP1 | - | 3 | 3 | no data | 10 | - | no data | 5.0 | no data | 3.0 | 2.0 | no data | 3.7 |
| PTPN11 | 8.51 | no data | no data | no data | - | 4.0 | 4.0 | 5.0 | 5.0 | 9.0 | 2.0 | 4.2 | 1.9 |
| ABL1 | - | - | - | no data | - | - | no data | - | - | - | - | no data | - |
| SMC3 | - | - | - | no data | - | 2.0 | no data | 2.0 | 4.0 | 2.0 | 4.0 | 2.1 | 1.9 |
| JAK2 | 0.71 | 1 | no data | no data | 10 | - | no data | no data | no data | no data | 2.0 | 1.1 | 5.6 |
| ETV6 | 1.43 | - | - | no data | 10 | 2.0 | no data | no data | no data | no data | 2.0 | 1.1 | 3.7 |
| PRPF40B | no data | - | - | no data | - | - | no data | no data | no data | no data | - | no data | 0.0 |
| MLL | no data | - | - | no data | - | 2.0 | no data | no data | no data | no data | - | 1.1 | 1.9 |
| RAD21 | 3.70 | - | - | no data | - | 2.0 | no data | 2.0 | 3.0 | 4.0 | 4.0 | 3.2 | 1.9 |
| GNAS | no data | - | - | no data | - | - | no data | no data | no data | no data | no data | - | 0.0 |
| CBL | 2.73 | 1 | 3 | no data | 10 | 3.0 | no data | 5.0 | 2.0 | 4.0 | 2.0 | no data | 5.6 |
| PHF6 | 3.05 | - | - | no data | 10 | 4.0 | 3.0 | 5.0 | no data | 1.0 | no data | 1.1 | 7.4 |
| SUZ12 | - | no data | no data | no data | - | - | no data | no data | no data | no data | - | - | 0.0 |
| CBLB | no data | - | no data | - | - | no data | no data | no data | no data | - | 2.1 | 0.0 | |
| MPL | no data | no data | no data | no data | 0 | - | no data | no data | no data | no data | no data | no data | 0.0 |
| SF3A1 | no data | - | no data | - | - | no data | no data | no data | no data | - | - | 0.0 | |
| SH2B3 | no data | no data | no data | no data | - | - | no data | no data | no data | no data | - | no data | 0.0 |
| U2AF2 | 0.13 | - | 14 | no data | - | - | no data | no data | no data | no data | - | no data | 0.0 |
| DAXX | - | - | - | no data | - | - | no data | - | - | - | - | - | 0.0 |
| EED | - | - | - | no data | - | - | no data | no data | no data | no data | - | - | 0.0 |
| RB1 | no data | - | - | no data | - | - | no data | - | - | - | - | no data | 0.0 |
| GATA1 | no data | - | - | no data | - | - | no data | no data | no data | no data | no data | no data | 0.0 |
| SF1 | no data | - | - | no data | - | - | no data | no data | no data | no data | no data | - | 0.0 |
| JAK1 | - | - | - | no data | - | - | no data | - | - | - | - | no data | 0.0 |
| CBFB | - | - | - | no data | - | - | no data | - | - | - | - | no data | 1.9 |
| CEBPA | 8.18 | 3 | 3 | 15 | 10 | 2.0 | 6.0 | 3.0 | 7.0 | 5.0 | 6.0 | 29.5 | 3.7 |
| Substance Group | Agent | Target | Ph | Patient Cohort | Schedule | ORR (%) | PFS (m) | OS (m) | A |
|---|---|---|---|---|---|---|---|---|---|
| FLT 3 Inhibitors | Midostaurin [61,62,63] | FLT3 (non mutated) | I | R/R or unfit | + AZA | 21 | NR | 6 | Y |
| FLT 3 mutated or WT | I | De novo | + CTx | 80 | NR | NR | |||
| FLT3 mutated or WT | II | R/R or unfit | M | 71 (mutated) 42 (WT) | NR | 4.3 | |||
| FLT3 mutated | III | De novo | + CTx | 59/53 | 26.7/15.5 | 74.7/25.6 | |||
| Quizartinib [64,65,66,67] | FLT3 mutated | I | R/R | M | 30 | NR | 3.5 | Y | |
| FLT3 mutated or WT | II | R/R | M | 47 | 2.2 | 6.7 | |||
| FLT3 mutated or WT | II | R/R | M | 74–77 | 3 | 6 | |||
| FLT3 mutated | III | R/R | M vs. CTx | 48 vs. 27 | 4 vs. 1.2 | 6.2 vs. 4.7 | |||
| Gilteritinib [68] | FLT3 (mutated) | I | R/R | M | 40 | 4.25 | 6.25 | Y | |
| Lestaurtinib [69] | FLT3 (mutated) | III | De novo | + CTx | 97 | 40 vs. 36% (NS) | 5 y OS 46 vs. 45% (NS) | N | |
| Sunitinib [70] | FLT 3 (mutated) | I/II | De novo elderly | + CTx | 59 | 12 | 18 | N | |
| Crenolanib [71,72,73,74,75] | FLT ITD and D835 | II | De novo | + CTx | 96 | NR | Nre | N | |
| II | De novo | + CTx | 83 | NR | Nre | ||||
| II | R/R | + CTx | 67 | NR | NR | ||||
| II | R/R | + CTx | 36 | NR | 9.25 | ||||
| II | R/R | M | 47 | 2 | 4.75 | ||||
| Sorafenib [76,77,78,79,80,81] | Multiple kinases | II | De novo | + CTx | 60 vs. 59 | 9 vs. 21 | Nre | N | |
| I | R/R | M | 10 | NR | NR | ||||
| I | After AlloTx in FLT3-ITD | M | NR | 85% at 12 Mo | 95% at 12 Mo | ||||
| I | R/R after AlloTx with FLT3-ITD | + AZA | 50 | NR | 322 days | ||||
| III | Maintenance After alloTx | M | NR | Nre vs. 30.9 | NR | ||||
| IDH Inhibitors | Enasidenib [82] | IDH 2 (mutated) | I/II | R/R | M | 40 | 6.4 | 9.3 | Y |
| Ivosidenib [83] | IDH 1 (mutated) | I | R/R | M | 41 | NR | NR | Y | |
| Bcl-2 Inhibitors | Venetoclax [84,85] | Bcl-2 | II | R/R or unfit | M | 19 | 2.5 | 4.7 | Y |
| Ib | unfit | + AZA or DAC | 73 | NR | 18 | ||||
| Obatoclax [86] | Bcl-2 family | I/II | unfit | M | 0 | NR | NR | N |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leisch, M.; Jansko, B.; Zaborsky, N.; Greil, R.; Pleyer, L. Next Generation Sequencing in AML—On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation? Cancers 2019, 11, 252. https://doi.org/10.3390/cancers11020252
Leisch M, Jansko B, Zaborsky N, Greil R, Pleyer L. Next Generation Sequencing in AML—On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation? Cancers. 2019; 11(2):252. https://doi.org/10.3390/cancers11020252
Chicago/Turabian StyleLeisch, Michael, Bettina Jansko, Nadja Zaborsky, Richard Greil, and Lisa Pleyer. 2019. "Next Generation Sequencing in AML—On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation?" Cancers 11, no. 2: 252. https://doi.org/10.3390/cancers11020252
APA StyleLeisch, M., Jansko, B., Zaborsky, N., Greil, R., & Pleyer, L. (2019). Next Generation Sequencing in AML—On the Way to Becoming a New Standard for Treatment Initiation and/or Modulation? Cancers, 11(2), 252. https://doi.org/10.3390/cancers11020252