Mouse-Derived Isograft (MDI) In Vivo Tumor Models I. Spontaneous sMDI Models: Characterization and Cancer Therapeutic Approaches

Abstract
1. Introduction
2. Results
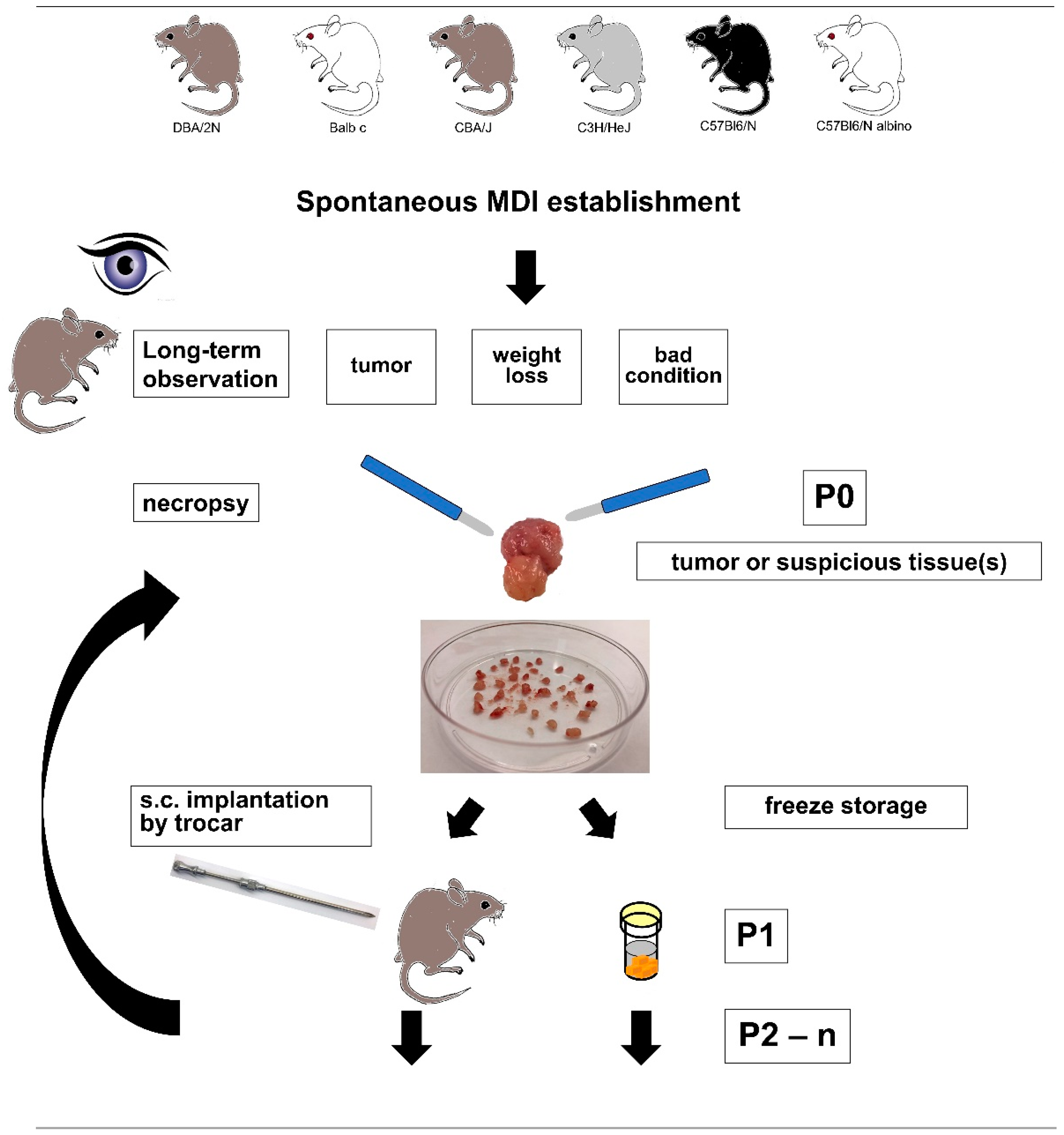
2.1. sMDI Establishment History
2.2. sMDI: Histological and Pathological Analysis
2.2.1. Hematopoietic Tumor Models
2.2.2. Non-Hematopoietic Tumor Models
2.3. sMDI: Take Rates and Growth Curves
2.4. sMDI: Flow Cytometric Analysis
2.5. sMDI/cMDI: Preliminary Results of RNA-Seq
2.6. sMDI: Efficacy Studies
3. Discussion
Future Outlook
4. Materials and Methods
4.1. Mouse Strains
4.2. sMDI: Establishment and Re-Transplantation
4.3. sMDI: Histological and Pathological Analysis
4.4. sMDI: Establishment and Efficacy Studies
4.5. Flow Cytometric Analysis of sMDI Infiltrating-Immune Cells
4.6. sMDI: RNA-Seq
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | adenocarcinoma |
| AP | appearance |
| bg | beige mutation |
| cMDI | carcinogen-induced mouse-derived isografts |
| CRL | Charles River Laboratories |
| DMSO | Dimethyl sulfoxide |
| FACS | fluorescence activated cell sorter |
| FPKM | fragments per kilobase million |
| HS/HAL | histiocytic sarcoma//histiocyte-associated lymphoma |
| ICPI | immune checkpoint inhibitors |
| IFN | interferon |
| IHC | immunohistochemistry |
| IRE | internal relative expression |
| LN | lymph node |
| MCA | 3-methylcholanthrene |
| mCTLA-4 | mouse cytotoxic T-lymphocyte-associated protein 4 |
| MDI | Mouse-derived isografts |
| cMDI | carcinogen-induced mouse-derived isograft |
| sMDI | spontaneous mouse-derived isograft |
| MDSC | myeloid-derived suppressor cells |
| MΦ | macrophage |
| MHC | major histocompatibility complex |
| M-MDSC | monocytic MDSC |
| MMR | macrophage mannose receptor |
| MNU | N-methyl-N-nitrosourea |
| mPD-1 | mouse programmed cell death protein 1 |
| NOD | non-obese diabetic |
| NOD mice | non-obese diabetic mice |
| NOS | not otherwise specified |
| NSG mice | NOD SCID gamma mice |
| PBS | phosphate-buffered saline |
| PDX | patient-derived xenografts |
| RNA | ribonucleic acid |
| RNA-Seq | RNA whole transcriptome shotgun sequencing |
| RPKM | reads per kilobase million |
| RRT | real running time |
| SCID | severe combined immunodeficiency disease |
| SCID mice | severe combined immunodeficiency mice |
| SCID/bg | SCID beige mutation |
| SCID/bg mice | severe combined immunodeficiency beige mice |
| TATA | tumor-associated transplantation antigens |
| Treg | regulatory T-cells |
| TTW | treatment time window |
| UKCCCR | United Kingdom Coordinating Committee of Cancer Research |
References
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-pd-l1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Ribas, A. Tumor immunotherapy directed at pd-1. N. Engl. J. Med. 2012, 366, 2517–2519. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-pd-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by ctla-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef]
- Prehn, R.T.; Main, J.M. Number of mouse histocompatibility genes involved in skin grafting from strain balb/can to strain dba/2. J. Natl. Cancer Inst. 1958, 20, 207–209. [Google Scholar] [PubMed]
- Snell, G.D. Antigenic differences between the sperm of different inbred strains of mice. Science 1944, 100, 272–273. [Google Scholar] [CrossRef] [PubMed]
- Strong, L.C. The establishment of the c(3)h inbred strain of mice for the study of spontaneous carcinoma of the mammary gland. Genetics 1935, 20, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; Loi, S.; Thomas, D.; Smyth, M.J. Mouse models of tumor immunotherapy. Adv. Immunol. 2016, 130, 1–24. [Google Scholar] [PubMed]
- Teicher, B.A. Tumor models for efficacy determination. Mol. Cancer Ther. 2006, 5, 2435–2443. [Google Scholar] [CrossRef]
- Hanahan, D.; Wagner, E.F.; Palmiter, R.D. The origins of oncomice: A history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 2007, 21, 2258–2270. [Google Scholar] [CrossRef] [PubMed]
- Prehn, R.T.; Main, J.M. Immunity to methylcholanthrene-induced sarcomas. J. Natl. Cancer Inst. 1957, 18, 769–778. [Google Scholar] [PubMed]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol. Cancer Res. 2008, 6, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve r&d productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Bosma, M.J.; Carroll, A.M. The scid mouse mutant: Definition, characterization, and potential uses. Annu. Rev. Immunol. 1991, 9, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.P. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet. Res. 1966, 8, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Rygaard, J.; Povlsen, C.O. Heterotransplantation of a human malignant tumour to “nude” mice. Acta Pathol. Microbiol. Scand. 1969, 77, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Long, J.C. Growth of cultured cells from patients with hodgkin’s disease and transplantation into nude mice. Proc. Natl. Acad. Sci. USA 1977, 74, 754–758. [Google Scholar] [CrossRef]
- Voskoglou-Nomikos, T.; Pater, J.L.; Seymour, L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 2003, 9, 4227–4239. [Google Scholar]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef]
- Williams, S.A.; Anderson, W.C.; Santaguida, M.T.; Dylla, S.J. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab. Investig. 2013, 93, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.R.; Wappett, M.; Beran, G.; Logie, A.; Delpuech, O.; Brown, H.; Boros, J.; Camp, N.J.; McEwen, R.; Mazzola, A.M.; et al. Whole transcriptome profiling of patient-derived xenograft models as a tool to identify both tumor and stromal specific biomarkers. Oncotarget 2016, 7, 20773–20787. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D.; Pham, K.; Vlada, A.C.; Sarosi, G.A.; Thomas, R.M.; Behrns, K.E.; Liu, C.; Hughes, S.J.; Wallet, S.M.; Trevino, J.G. Patient-derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements. Am. J. Pathol. 2015, 185, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Merino-Trigo, A.; Lacroix, L.; Pocard, M.; Goere, D.; Mariani, P.; Landron, S.; Bigot, L.; Nemati, F.; Dartigues, P.; et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin. Cancer Res. 2012, 18, 5314–5328. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, V.E.; Allaj, V.; Gardner, E.E.; Poirier, J.T.; Rudin, C.M. Quantitation of murine stroma and selective purification of the human tumor component of patient-derived xenografts for genomic analysis. PLoS ONE 2016, 11, e0160587. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.N.; Higgins, G.S.; Brown, J.M.; Baumann, M.; Kirsch, D.G.; Willers, H.; Prasanna, P.G.; Dewhirst, M.W.; Bernhard, E.J.; Ahmed, M.M. Improving the predictive value of preclinical studies in support of radiotherapy clinical trials. Clin. Cancer Res. 2016, 22, 3138–3147. [Google Scholar] [CrossRef] [PubMed]
- Unger, C.; Kramer, N.; Walzl, A.; Scherzer, M.; Hengstschlager, M.; Dolznig, H. Modeling human carcinomas: Physiologically relevant 3d models to improve anti-cancer drug development. Adv. Drug Deliv. Rev. 2014, 79–80, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Nicosia, S.V.; Dave, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2014, 344, 1–12. [Google Scholar] [CrossRef]
- Pimiento, J.M.; Larkin, E.M.; Smalley, K.S.; Wiersma, G.L.; Monks, N.R.; Fedorenko, I.V.; Peterson, C.A.; Nickoloff, B.J. Melanoma genotypes and phenotypes get personal. Lab. Investig. 2013, 93, 858–867. [Google Scholar] [CrossRef]
- Reyal, F.; Guyader, C.; Decraene, C.; Lucchesi, C.; Auger, N.; Assayag, F.; De Plater, L.; Gentien, D.; Poupon, M.F.; Cottu, P.; et al. Molecular profiling of patient-derived breast cancer xenografts. Breast Cancer Res. 2012, 14, R11. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Hidalgo, M.; Kung, A.L. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 2015, 15, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Racki, W.J.; Covassin, L.; Brehm, M.; Pino, S.; Ignotz, R.; Dunn, R.; Laning, J.; Graves, S.K.; Rossini, A.A.; Shultz, L.D.; et al. Nod-scid il2rgamma(null) mouse model of human skin transplantation and allograft rejection. Transplantation 2010, 89, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Hannon, G.J. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Herndler-Brandstetter, D.; Shan, L.; Yao, Y.; Stecher, C.; Plajer, V.; Lietzenmayer, M.; Strowig, T.; de Zoete, M.R.; Palm, N.W.; Chen, J.; et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9626–E9634. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Song, S.J.; Wu, Z.Z.; Wu, W.; Cui, X.Y.; Chen, J.N.; Zeng, M.S.; Su, S.C. Epstein-barr virus-induced vegf and gm-csf drive nasopharyngeal carcinoma metastasis via recruitment and activation of macrophages. Cancer Res. 2017, 77, 3591–3604. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of pd-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef]
- Fu, J.; Sen, R.; Masica, D.L.; Karchin, R.; Pardoll, D.; Walter, V.; Hayes, D.N.; Chung, C.H.; Kim, Y.J. Autologous reconstitution of human cancer and immune system in vivo. Oncotarget 2017, 8, 2053–2068. [Google Scholar] [CrossRef] [PubMed]
- Tormo, A.; Khodayarian, F.; Cui, Y.; Al-Chami, E.; Kanjarawi, R.; Noe, B.; Wang, H.; Rafei, M. Interleukin-21 promotes thymopoiesis recovery following hematopoietic stem cell transplantation. J. Hematol. Oncol. 2017, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Sundarasetty, B.; Volk, V.; Theobald, S.J.; Rittinghausen, S.; Schaudien, D.; Neuhaus, V.; Figueiredo, C.; Schneider, A.; Gerasch, L.; Mucci, A.; et al. Human effector memory t helper cells engage with mouse macrophages and cause graft-versus-host-like pathology in skin of humanized mice used in a nonclinical immunization study. Am. J. Pathol. 2017, 187, 1380–1398. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.; Christoforidou, Z.; Lewis, M.C. The evolutionary basis for differences between the immune systems of man, mouse, pig and ruminants. Vet. Immunol. Immunopathol. 2013, 152, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Shay, T.; Jojic, V.; Zuk, O.; Rothamel, K.; Puyraimond-Zemmour, D.; Feng, T.; Wakamatsu, E.; Benoist, C.; Koller, D.; Regev, A.; et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc. Natl. Acad. Sci. USA 2013, 110, 2946–2951. [Google Scholar] [CrossRef] [PubMed]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef] [PubMed]
- Haley, P.J. Species differences in the structure and function of the immune system. Toxicology 2003, 188, 49–71. [Google Scholar] [CrossRef]
- Dranoff, G. Experimental mouse tumour models: What can be learnt about human cancer immunology? Nat. Rev. Immunol. 2012, 12, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.E.; Junttila, M.R.; de Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef]
- Belzung, C.; Barreau, S. Differences in drug-induced place conditioning between balb/c and c57bl/6 mice. Pharmacol. Biochem. Behav. 2000, 65, 419–423. [Google Scholar] [CrossRef]
- Chen, X.; Oppenheim, J.J.; Howard, O.M. Balb/c mice have more cd4+cd25+ t regulatory cells and show greater susceptibility to suppression of their cd4+cd25- responder t cells than c57bl/6 mice. J. Leukoc. Biol. 2005, 78, 114–121. [Google Scholar] [CrossRef]
- Melgar, S.; Karlsson, A.; Michaelsson, E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in c57bl/6 but not in balb/c mice: Correlation between symptoms and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.F.; Nebane-Ambe, D.L.; Chhabra, A.; Parnell, S.A.; Zhao, Y.; Alard, P.; Kosiewicz, M.M. Decreased frequencies of cd4+cd25+foxp3+ cells and the potent cd103+ subset in peripheral lymph nodes correlate with autoimmune disease predisposition in some strains of mice. Autoimmunity 2011, 44, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Petkov, P.M.; Ding, Y.; Cassell, M.A.; Zhang, W.; Wagner, G.; Sargent, E.E.; Asquith, S.; Crew, V.; Johnson, K.A.; Robinson, P.; et al. An efficient snp system for mouse genome scanning and elucidating strain relationships. Genome Res. 2004, 14, 1806–1811. [Google Scholar] [CrossRef]
- Watanabe, H.; Numata, K.; Ito, T.; Takagi, K.; Matsukawa, A. Innate immune response in th1- and th2-dominant mouse strains. Shock 2004, 22, 460–466. [Google Scholar] [CrossRef]
- Kuroda, E.; Sugiura, T.; Zeki, K.; Yoshida, Y.; Yamashita, U. Sensitivity difference to the suppressive effect of prostaglandin e2 among mouse strains: A possible mechanism to polarize th2 type response in balb/c mice. J. Immunol. 2000, 164, 2386–2395. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hosiawa, K.A.; Min, W.; Yang, J.; Zhang, X.; Garcia, B.; Ichim, T.E.; Zhou, D.; Lian, D.; Kelvin, D.J.; et al. Cytokines regulate the pattern of rejection and susceptibility to cyclosporine therapy in different mouse recipient strains after cardiac allografting. J. Immunol. 2003, 171, 3823–3836. [Google Scholar] [CrossRef] [PubMed]
- Sellers, R.S.; Clifford, C.B.; Treuting, P.M.; Brayton, C. Immunological variation between inbred laboratory mouse strains: Points to consider in phenotyping genetically immunomodified mice. Vet. Pathol. 2012, 49, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Jovicic, N.; Jeftic, I.; Jovanovic, I.; Radosavljevic, G.; Arsenijevic, N.; Lukic, M.L.; Pejnovic, N. Differential immunometabolic phenotype in th1 and th2 dominant mouse strains in response to high-fat feeding. PLoS ONE 2015, 10, e0134089. [Google Scholar] [CrossRef] [PubMed]
- Beshay, J.; Jantscheff, P.; Lemarchand, T.; Obodozie, C.; Schächtele, C.; Weber, H. New mouse-derived isograft (mdi) tumor models ii. Carcinogen-induced cmdi models for immune as well as other therapeutic approaches. Cancers 2019, 11, 242. [Google Scholar] [CrossRef]
- Hao, X.; Fredrickson, T.N.; Chattopadhyay, S.K.; Han, W.; Qi, C.F.; Wang, Z.; Ward, J.M.; Hartley, J.W.; Morse, H.C., 3rd. The histopathologic and molecular basis for the diagnosis of histiocytic sarcoma and histiocyte-associated lymphoma of mice. Vet. Pathol. 2010, 47, 434–445. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- METD Database: Whereby XX Was Taken for the Respective Gene Name. Available online: https://www.ncbi.nlm.nih.gov/gene/?term=XX+mus+musculus (accessed on 16 February 2019).
- Juacaba, S.F.; Horak, E.; Price, J.E.; Tarin, D. Tumor cell dissemination patterns and metastasis of murine mammary carcinoma. Cancer Res. 1989, 49, 570–575. [Google Scholar] [PubMed]
- McFarland, V.W.; Mora, P.T.; Schultz, A.; Pancake, S. Cell properties after repeated transplantation of spontaneously and of sv40 virus transformed mouse cell lines. I. Growth in culture. J. Cell Physiol. 1975, 85, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Shepherd, F.; Gropp, T. Enhancement of the tumorigenicity of preneoplastic mammary nodule lines by enzymatic dissociation. J. Natl. Cancer Inst. 1978, 60, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Erenus, N.; Muller, J.H. Versuche uber Zuchtung von Tumorzellen aus Ascites. Gynaecologia 1948, 126, 13–18. [Google Scholar] [PubMed]
- Nadel, E.M.; Greenberg, J. Synergistic inhibitory action of a-methopterin and a diaminopyrimidine upon leukemia l1210 in mice. Cancer Res. 1953, 13, 865–868. [Google Scholar] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Drake, W.P.; Pendergrast, W.J., Jr.; Kramer, R.E.; Mardiney, M.R., Jr. Enhancement of spontaneous c3h/hej mammary tumorigenesis by long-term polyadenylic-polyuridylic acid therapy. Cancer Res. 1975, 35, 3051–3053. [Google Scholar]
- Mahler, J.F.; Stokes, W.; Mann, P.C.; Takaoka, M.; Maronpot, R.R. Spontaneous lesions in aging fvb/n mice. Toxicol. Pathol. 1996, 24, 710–716. [Google Scholar] [CrossRef]
- Schechter, J.E.; Felicio, L.S.; Nelson, J.F.; Finch, C.E. Pituitary tumorigenesis in aging female c57bl/6j mice: A light and electron microscopic study. Anat. Rec. 1981, 199, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Popovich, I.G.; Zabezhinski, M.A.; Egormin, P.A.; Yurova, M.N.; Semenchenko, A.V.; Tyndyk, M.L.; Panchenko, A.V.; Trashkov, A.P.; Vasiliev, A.G.; et al. Sex differences in aging, life span and spontaneous tumorigenesis in 129/sv mice neonatally exposed to metformin. Cell Cycle 2015, 14, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Baldrick, P.; Reeve, L. Carcinogenicity evaluation: Comparison of tumor data from dual control groups in the cd-1 mouse. Toxicol. Pathol. 2007, 35, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Prochazka, M.; Gaskins, H.R.; Shultz, L.D.; Leiter, E.H. The nonobese diabetic scid mouse: Model for spontaneous thymomagenesis associated with immunodeficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 3290–3294. [Google Scholar] [CrossRef] [PubMed]
- Nci Dictionary of Cancer Terms: Solid Tumor. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/solid-tumor (accessed on 16 February 2019).
- Koren, S.; Bentires-Alj, M. Breast tumor heterogeneity: Source of fitness, hurdle for therapy. Mol. Cell 2015, 60, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Jantscheff, P.; Esser, N.; Graeser, R.; Ziroli, V.; Kluth, J.; Unger, C.; Massing, U. Liposomal gemcitabine (gemlip)-efficient drug against hormone-refractory du145 and pc-3 prostate cancer xenografts. Prostate 2009, 69, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Jantscheff, P.; Ziroli, V.; Esser, N.; Graeser, R.; Kluth, J.; Sukolinskaya, A.; Taylor, L.A.; Unger, C.; Massing, U. Anti-metastatic effects of liposomal gemcitabine in a human orthotopic lncap prostate cancer xenograft model. Clin. Exp. Metastasis 2009, 26, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Graeser, R.; Bornmann, C.; Esser, N.; Ziroli, V.; Jantscheff, P.; Unger, C.; Hopt, U.T.; Schaechtele, C.; von Dobschuetz, E.; Massing, U. Antimetastatic effects of liposomal gemcitabine and empty liposomes in an orthotopic mouse model of pancreatic cancer. Pancreas 2009, 38, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Medscape: Gemzar-gemcitabine. 2018. Available online: https://reference.medscape.com/drug/infugem-gemzar-gemcitabine-342218 (accessed on 16 February 2019).
- Jantscheff, P.; Schaefer-Obodozie, C.; Moor, S.; Weber, H. Pd-1, anti-pd-l1 and anti-ctla-4 checkpoint inhibitor treatment leads to different responses in syngeneic tumor models. [abstract]. In Proceedings of the 107th Annual Meeting of the American Association for Cancer Research, New Orleans, LA, USA, 16–20 April 2016; AACR: Philadelphia, PA, USA, 2016; p. 76, abstract nr 3216. [Google Scholar]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Kimura, Y.; Sumiyoshi, M. Antitumor and antimetastatic actions of dihydroxycoumarins (esculetin or fraxetin) through the inhibition of m2 macrophage differentiation in tumor-associated macrophages and/or g1 arrest in tumor cells. Eur. J. Pharmacol. 2015, 746, 115–125. [Google Scholar] [CrossRef]
- Sugimoto, M.; Mitsunaga, S.; Yoshikawa, K.; Kato, Y.; Gotohda, N.; Takahashi, S.; Konishi, M.; Ikeda, M.; Kojima, M.; Ochiai, A.; et al. Prognostic impact of m2 macrophages at neural invasion in patients with invasive ductal carcinoma of the pancreas. Eur. J. Cancer 2014, 50, 1900–1908. [Google Scholar] [CrossRef] [PubMed]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: Relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; He, X.J.; Wang, H.J.; Xia, Y.J.; Wang, S.L.; Ye, Z.Y.; Tao, H.Q. Interaction of coagulation factors and tumor-associated macrophages mediates migration and invasion of gastric cancer. Cancer Sci. 2011, 102, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Mitsunaga, S.; Kinoshita, T.; Konishi, M.; Takahashi, S.; Gotohda, N.; Kato, Y.; Aizawa, M.; Ochiai, A. Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 2012, 103, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Udayanga, K.G.; Totsuka, N.; Weinberg, J.B.; Nunez, G.; Shibuya, A. Gut dysbiosis promotes m2 macrophage polarization and allergic airway inflammation via fungi-induced pge(2). Cell Host Microbe 2014, 15, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Ho-Yen, C.M.; Jones, J.L.; Kermorgant, S. The clinical and functional significance of c-met in breast cancer: A review. Breast Cancer Res. 2015, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Rohan, R.M.; Fernandez, A.; Udagawa, T.; Yuan, J.; D’Amato, R.J. Genetic heterogeneity of angiogenesis in mice. FASEB J. 2000, 14, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Marmiroli, P.; Riva, B.; Pozzi, E.; Ballarini, E.; Lim, D.; Chiorazzi, A.; Meregalli, C.; Distasi, C.; Renn, C.L.; Semperboni, S.; et al. Susceptibility of different mouse strains to oxaliplatin peripheral neurotoxicity: Phenotypic and genotypic insights. PLoS ONE 2017, 12, e0186250. [Google Scholar] [CrossRef] [PubMed]
- Vikis, H.G.; Jackson, E.N.; Krupnick, A.S.; Franklin, A.; Gelman, A.E.; Chen, Q.; Piwnica-Worms, D.; You, M. Strain-specific susceptibility for pulmonary metastasis of sarcoma 180 cells in inbred mice. Cancer Res. 2010, 70, 4859–4867. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Balmain, A.; Hickman, J.A.; McNally, N.J.; Rohas, A.M.; Mitchison, N.A.; Pierrepoint, C.G.; Raymond, R.; Rowlatt, C.; Stephens, T.C.; et al. Ukcccr guidelines for the welfare of animals in experimental neoplasia. Lab. Anim. 1988, 22, 195–201. [Google Scholar] [CrossRef]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An in vivo platform for translational drug development in pancreatic cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Mangsbo, S.M.; Sandin, L.C.; Anger, K.; Korman, A.J.; Loskog, A.; Totterman, T.H. Enhanced tumor eradication by combining ctla-4 or pd-1 blockade with cpg therapy. J. Immunother. 2010, 33, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Pachter, L. Models for Transcript Quantification from RNA-Seq. arXiv, 2011; arXiv:1104.3889v2. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spontaneous MDI | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor Name | JA-0009 | JA-0011 | JA-0013 | JA-0017 | JA-0018 | JA-0023 | JA-0032 | JA-0034 | JA-0021 | |
| (back) | (kidney) | |||||||||
| Hematopoietic (y/n) | no | yes (?) | yes | no | yes | no | no | no | yes | yes |
| Histopathological Diagnosis | low differentiated ADC | HS or HAL | malignant lymphoma | ADC | malignant lymphoma | mammary invasive ADC | clear cell-like ADC of kidney | papillary ADC lung | malignant lymphoma | malignant lymphoma |
| Mouse Strain | DBA/2N | DBA/2N | C57BL/6N albino | CBA/J | CBA/J | C3H/HeJ | C3H/HeJ | BALB/c | C57BL/6N albino | thymus (THY) SCID/bg 1205-16 |
| Sex | ♀ | ♀ | ♀ | ♀ | ♀ | ♀ | ♀ | ♂ | ♂ | ♀ |
| Estimated Growth Time (In Days) | 21–23 | 27–72 problem: liver-/splenomegaly | 45 | 81 to ≥158 | 60 | 29–45 | 60 | 45–59 | 45 | 24 |
| Included Animals | growth curve (12) * | frozen/directly (12/10) *** Pool § | frozen (11) ** | growth curve (12) | frozen/directly (3/3) | growth curve (12) | frozen/directly (3/4) | growth curve (12) | frozen (12) only gut | frozen/directly (3/6) only SCID/bg |
| Take Rate | 92% 11/12 | 63% 14/22 & | 100% 11/11 | 75% 9/12 | 100% 6/6 | 100% 12/12 | 71% 5/7 | 66% 8/12 | 92% 11/12 | 78% 7/9 |
| Appearance (AP) Day | 6–7 | nd | nd | 49–56 | nd | 13–20 | nd | 31 - 41 | nd | nd |
| Real Running Time (RRT) Day (% Alive Animals) | 21 (75%) 23 (67%) | nd | nd | 81 (58%) 105 (50%) | nd | 29 (92%) 45 (42%) | nd | 45 (67%) 59 (42%) | nd | nd has to be done in H-2d × H-2b hybrid mouse |
| Mouse Strain | CRL Nomenclature | Sex | H-2 Haplotype | Tumor Growth | |
|---|---|---|---|---|---|
| n | (n) | ||||
| Black 6 | C57BL/6NCrl | ♂ | H-2b | - | (1) |
| ♀ | |||||
| Black 6 albino | C57BL/6N-Tyrc-Brd/BrdCrCrl | ♂ | H-2b | 1 | (1) |
| ♀ | 1 | (1) | |||
| Balb/c | BALB/cAnNCrl | ♂ | H-2d | 1 | (2) |
| ♀ | - | (1) | |||
| CBA/J | CBA/J-Pde6brd1 | ♀ | H-2k | 2 | - |
| DBA/2 | DBA/2NCrl | ♀ | H-2d | 2 | - |
| C3H/HeJ | C3H/HeJ-Pde6brd1 | ♀ | H-2k | 1 | (1) |
| SCID beige | CB17.Cg-PrkdcscidLystbg/Crl | ♀ | H-2d × H-2b | 1 * | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jantscheff, P.; Beshay, J.; Lemarchand, T.; Obodozie, C.; Schächtele, C.; Weber, H. Mouse-Derived Isograft (MDI) In Vivo Tumor Models I. Spontaneous sMDI Models: Characterization and Cancer Therapeutic Approaches. Cancers 2019, 11, 244. https://doi.org/10.3390/cancers11020244
Jantscheff P, Beshay J, Lemarchand T, Obodozie C, Schächtele C, Weber H. Mouse-Derived Isograft (MDI) In Vivo Tumor Models I. Spontaneous sMDI Models: Characterization and Cancer Therapeutic Approaches. Cancers. 2019; 11(2):244. https://doi.org/10.3390/cancers11020244
Chicago/Turabian StyleJantscheff, Peter, Janette Beshay, Thomas Lemarchand, Cynthia Obodozie, Christoph Schächtele, and Holger Weber. 2019. "Mouse-Derived Isograft (MDI) In Vivo Tumor Models I. Spontaneous sMDI Models: Characterization and Cancer Therapeutic Approaches" Cancers 11, no. 2: 244. https://doi.org/10.3390/cancers11020244
APA StyleJantscheff, P., Beshay, J., Lemarchand, T., Obodozie, C., Schächtele, C., & Weber, H. (2019). Mouse-Derived Isograft (MDI) In Vivo Tumor Models I. Spontaneous sMDI Models: Characterization and Cancer Therapeutic Approaches. Cancers, 11(2), 244. https://doi.org/10.3390/cancers11020244