The Diverse Consequences of FOXC1 Deregulation in Cancer


Abstract
:1. FOXC1 Structure and Function
2. FOXC1 in Tissue Development, Homeostasis and Disease
2.1. FOXC1 and Development
2.2. FOXC1 Contributes to the Maintenance of Adult Stem and Progenitor Cell Compartments
2.3. FOXC1 Overexpression is a Frequent and Functional Event in Cancer
3. FOXC1 Action Across the Hallmarks of Cancer
3.1. FOXC1 Enhances Cancer Cell Proliferation and Survival in Diverse Cancer Types
3.2. FOXC1 As a Putative Regulator of Cancer Stem Cell Function
3.3. FOXC1 Expression Enhances the Adaptation to Tumor Hypoxia
3.4. FOXC1 Activates Genes Heralding the Epithelial-Mesenchymal Transition and Tumor Migration
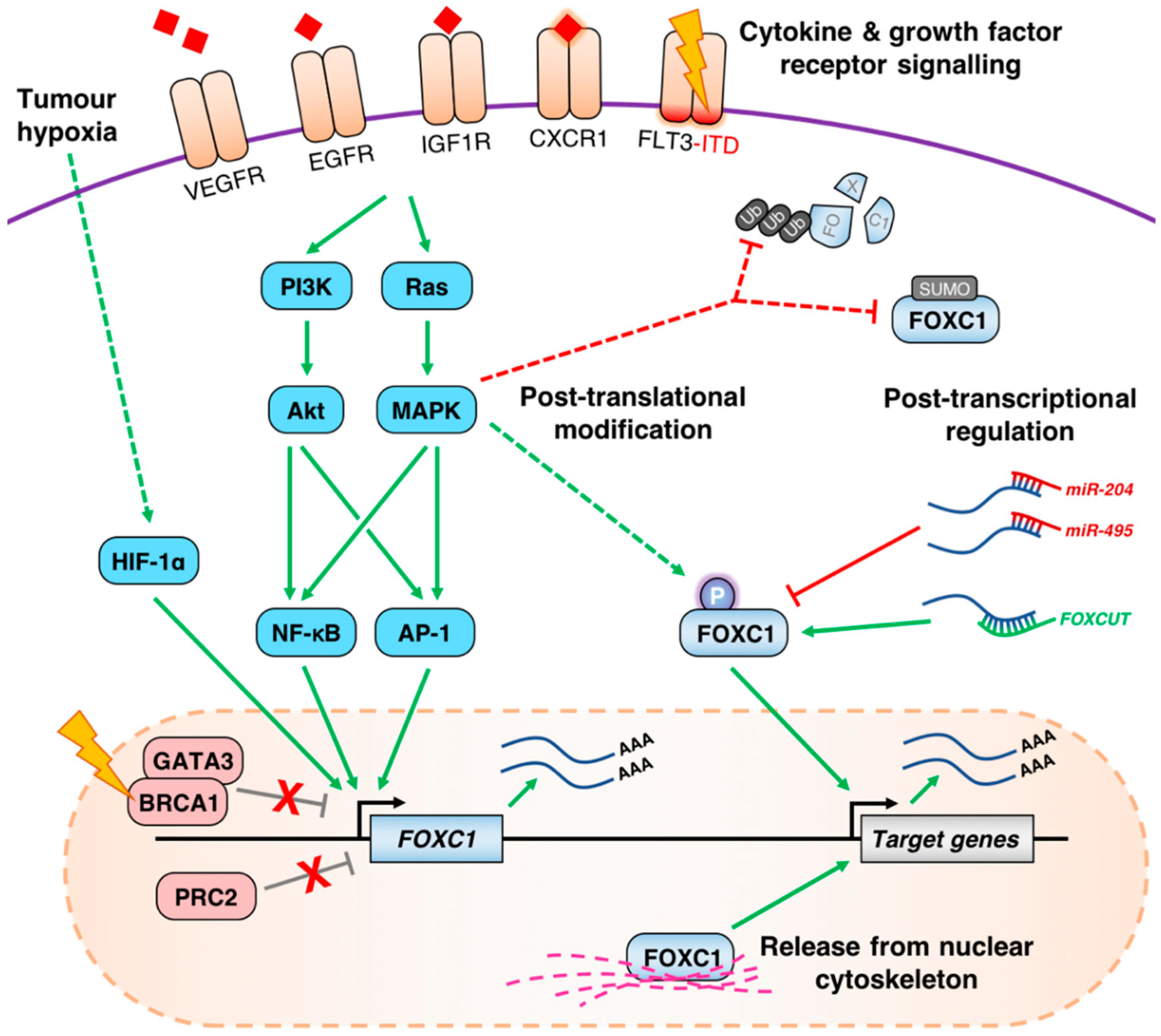
4. Mechanisms of FOXC1 Deregulation in Cancer
4.1. Aberrant de-Repression of the FOXC1 Locus
4.2. Growth Factor Receptor Signaling Pathways Influence the Expression of FOXC1
4.3. Post-Transcriptional and Post-Translational Regulation of FOXC1 in Cancer
5. The Utility of FOXC1 as a Cancer Biomarker
6. Conclusions, Current Limitations and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Larsson, C.; Hellqvist, M.; Pierrou, S.; White, I.; Enerbäck, S.; Carlsson, P. Chromosomal localization of six human forkhead genes, FREAC-1 (FKHL5), -3 (FKHL7), -4 (FKHL8), -5 (FKHL9), -6 (FKHL10), and -8 (FKHL12). Genomics 1995, 30, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Kume, T.; Deng, K.Y.; Winfrey, V.; Gould, D.B.; Walter, M.A.; Hogan, B.L. The Forkhead/Winged Helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 1998, 93, 985–996. [Google Scholar] [CrossRef]
- Jackson, B.C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse forkhead box (FOX) gene families. Hum. Genom. 2010, 4, 345–352. [Google Scholar]
- Pierrou, S.; Hellqvist, M.; Samuelsson, L.; Enerbäck, S.; Carlsson, P. Cloning and characterization of seven human forkhead proteins: Binding site specificity and DNA bending. EMBO J. 1994, 13, 5002–5012. [Google Scholar] [CrossRef] [PubMed]
- Seifi, M.; Walter, M.A. Axenfeld-Rieger syndrome. Clin. Genet. 2018, 93, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.A.; McPherson, C.E.; Bossard, P.; Stevens, K.; Cherian, S.; Shim, E.Y.; Clark, K.L.; Burley, S.K.; Zaret, K.S. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998, 17, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yusuf, I.; Andersen, H.M.; Fruman, D.A. FOXO transcription factors cooperate with delta EF1 to activate growth suppressive genes in B lymphocytes. J. Immunol. 2006, 176, 2711–2721. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, I.; Zaret, K.S.; Santisteban, P. The forkhead factor FoxE1 binds to the thyroperoxidase promoter during thyroid cell differentiation and modifies compacted chromatin structure. Mol. Cell. Biol. 2007, 27, 7302–7314. [Google Scholar] [CrossRef]
- Sekiya, T.; Muthurajan, U.M.; Luger, K.; Tulin, A.V.; Zaret, K.S. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009, 23, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Caravaca, J.M.; Donahue, G.; Becker, J.S.; He, X.; Vinson, C.; Zaret, K.S. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013, 27, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Iwafuchi-Doi, M.; Donahue, G.; Kakumanu, A.; Watts, J.A.; Mahony, S.; Pugh, B.F.; Lee, D.; Kaestner, K.H.; Zaret, K.S. The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Mol. Cell 2016, 62, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Mango, S.E. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr. Opin. Genet. Dev. 2016, 37, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a critical determinant of Estrogen Receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Berry, F.B.; Saleem, R.A.; Walter, M.A. FOXC1 Transcriptional Regulation Is Mediated by N- and C-terminal Activation Domains and Contains a Phosphorylated Transcriptional Inhibitory Domain. J. Biol. Chem. 2002, 277, 10292–10297. [Google Scholar] [CrossRef]
- Berry, F.B.; Mirzayans, F.; Walter, M.A. Regulation of FOXC1 Stability and Transcriptional Activity by an Epidermal Growth Factor-activated Mitogen-activated Protein Kinase Signaling Cascade. J. Biol. Chem. 2006, 281, 10098–10104. [Google Scholar] [CrossRef]
- Danciu, T.E.; Chupreta, S.; Cruz, O.; Fox, J.E.; Whitman, M.; Iñiguez-Lluhí, J.A. Small Ubiquitin-like Modifier (SUMO) Modification Mediates Function of the Inhibitory Domains of Developmental Regulators FOXC1 and FOXC2. J. Biol. Chem. 2012, 287, 18318–18329. [Google Scholar] [CrossRef]
- Chalamalasetty, R.B.; Garriock, R.J.; Dunty, W.C.; Kennedy, M.W.; Jailwala, P.; Si, H.; Yamaguchi, T.P. Mesogenin 1 is a master regulator of paraxial presomitic mesoderm differentiation. Development 2014, 141, 4285–4297. [Google Scholar] [CrossRef]
- Sun, J.; Ishii, M.; Ting, M.C.; Maxson, R. Foxc1 controls the growth of the murine frontal bone rudiment by direct regulation of a Bmp response threshold of Msx2. Dev. Camb. Engl. 2013, 140, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Hata, K.; Takashima, R.; Ono, K.; Nakamura, E.; Takahata, Y.; Murakami, T.; Iseki, S.; Takano-Yamamoto, T.; Nishimura, R.; et al. The transcription factor Foxc1 is necessary for Ihh–Gli2-regulated endochondral ossification. Nat. Commun. 2015, 6, 6653. [Google Scholar] [CrossRef] [PubMed]
- Kume, T.; Deng, K.; Hogan, B.L. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Dev. Camb. Engl. 2000, 127, 1387–1395. [Google Scholar]
- Kume, T.; Jiang, H.; Topczewska, J.M.; Hogan, B.L. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 2001, 15, 2470–2482. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Chen, L.; Liu, W.; Zhao, D.; Schultz, K.M.; Sasman, A.; Liu, T.; Zhang, H.F.; Gage, P.J.; Kume, T. Foxc1 and Foxc2 in the Neural Crest Are Required for Ocular Anterior Segment Development. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Saleem, R.A.; Banerjee-Basu, S.; Berry, F.B.; Baxevanis, A.D.; Walter, M.A. Analyses of the effects that disease-causing missense mutations have on the structure and function of the winged-helix protein FOXC1. Am. J. Hum. Genet. 2001, 68, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Saleem, R.A.; Banerjee-Basu, S.; Berry, F.B.; Baxevanis, A.D.; Walter, M.A. Structural and functional analyses of disease-causing missense mutations in the forkhead domain of FOXC1. Hum. Mol. Genet. 2003, 12, 2993–3005. [Google Scholar] [CrossRef]
- Saleem, R.A.; Banerjee-Basu, S.; Murphy, T.C.; Baxevanis, A.; Walter, M.A. Essential structural and functional determinants within the forkhead domain of FOXC1. Nucleic Acids Res. 2004, 32, 4182–4193. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Lehmann, O.J.; Hudgins, L.; Chizhikov, V.V.; Bassuk, A.G.; Ades, L.C.; Krantz, I.D.; Dobyns, W.B.; Millen, K.J. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat. Genet. 2009, 41, 1037–1042. [Google Scholar] [CrossRef]
- Haldipur, P.; Dang, D.; Aldinger, K.A.; Janson, O.K.; Guimiot, F.; Adle-Biasette, H.; Dobyns, W.B.; Siebert, J.R.; Russo, R.; Millen, K.J. Phenotypic outcomes in Mouse and Human Foxc1 dependent Dandy-Walker cerebellar malformation suggest shared mechanisms. eLife 2017, 6, e20898. [Google Scholar] [CrossRef]
- French, C.R.; Seshadri, S.; Destefano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J. Clin. Investig. 2014, 124, 4877–4881. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Siegenthaler, J.A.; Dowell, R.D.; Yi, R. Foxc1 reinforces quiescence in self-renewing hair follicle stem cells. Science 2016, 351, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Lay, K.; Kume, T.; Fuchs, E. FOXC1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proc. Natl. Acad. Sci. USA 2016, 113, E1506–E1515. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Cao, P.; Wang, C.; Huang, D.; Lian, M.; Song, Y.; Yin, W.; Zheng, K.; Gu, Z.; Gu, Y.; et al. The Forkhead Box C1, a Novel Negative Regulator of Osteogenesis, Plays a Crucial Role in Odontogenic Differentiation of Dental Pulp Stem Cells. Cell. Reprogram. 2018, 20, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Bloushtain-Qimron, N.; Yao, J.; Snyder, E.L.; Shipitsin, M.; Campbell, L.L.; Mani, S.A.; Hu, M.; Chen, H.; Ustyansky, V.; Antosiewicz, J.E.; et al. Cell type-specific DNA methylation patterns in the human breast. Proc. Natl. Acad. Sci. USA 2008, 105, 14076–14081. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, G.M.; Sizemore, S.T.; Pal, B.; Booth, C.N.; Seachrist, D.D.; Abdul-Karim, F.W.; Kume, T.; Keri, R.A. FOXC1 is enriched in the mammary luminal progenitor population, but is not necessary for mouse mammary ductal morphogenesis. Biol. Reprod. 2013, 89, 10. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.J.H.F.; Kannan, N.; Pellacani, D.; Eaves, C.J. Mass Cytometric Analysis Reveals Viable Activated Caspase-3+ Luminal Progenitors in the Normal Adult Human Mammary Gland. Cell. Rep. 2017, 21, 1116–1126. [Google Scholar] [CrossRef]
- Gao, B.; Qu, Y.; Han, B.; Nagaoka, Y.; Katsumata, M.; Deng, N.; Bose, S.; Jin, L.; Giuliano, A.E.; Cui, X. Inhibition of lobuloalveolar development by FOXC1 overexpression in the mouse mammary gland. Sci. Rep. 2017, 7, 14017. [Google Scholar] [CrossRef]
- Omatsu, Y.; Seike, M.; Sugiyama, T.; Kume, T.; Nagasawa, T. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature 2014, 508, 536–540. [Google Scholar] [CrossRef]
- Somerville, T.D.D.; Wiseman, D.H.; Spencer, G.J.; Huang, X.; Lynch, J.T.; Leong, H.S.; Williams, E.L.; Cheesman, E.; Somervaille, T.C. Frequent Derepression of the Mesenchymal Transcription Factor Gene FOXC1 in Acute Myeloid Leukemia. Cancer Cell 2015, 28, 329–342. [Google Scholar] [CrossRef]
- Swaminathan, M.; Jensen, T.W.; Ray, T.; Andruska, N.D.; Shah, A.; Egner, J.R.; Ray, P.S. FOXC1 Expression in Acute Myeloid Leukemia: Potential Predictor of Disease Relapse and/or Refractory Disease. Blood 2016, 128, 5260. [Google Scholar]
- Ray, P.S.; Wang, J.; Qu, Y.; Sim, M.S.; Shamonki, J.; Bagaria, S.P.; Ye, X.; Liu, B.; Elashoff, D.; Hoon, D.S.; et al. FOXC1 is a potential prognostic biomarker with functional significance in basal-like breast cancer. Cancer Res. 2010, 70, 3870–3876. [Google Scholar] [CrossRef] [PubMed]
- Hirukawa, A.; Smith, H.W.; Zuo, D.; Dufour, C.R.; Savage, P.; Bertos, N.; Johnson, R.M.; Bui, T.; Bourque, G.; Basik, M.; et al. Targeting EZH2 reactivates a breast cancer subtype-specific anti-metastatic transcriptional program. Nat. Commun. 2018, 9, 2547. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wei, P.; Wu, J.; Zhang, M.; Li, G.; Li, Y.; Xu, Y.; Li, X.; Xie, D.; Cai, S.; et al. The FOXC1/FBP1 signaling axis promotes colorectal cancer proliferation by enhancing the Warburg effect. Oncogene 2018, 38, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Z.; Li, X.; Chen, J.; Wang, G.; Tian, Z.; Qian, M.; Chen, Z.; Guo, H.; Tang, G.; et al. Forkhead box C1 promotes colorectal cancer metastasis through transactivating ITGA7 and FGFR4 expression. Oncogene 2018, 37, 5477–5491. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Huang, Z.; Fan, Y.; He, L.; Ye, M.; Shi, K.; Ji, B.; Huang, J.; Wang, Y.; Li, Q. FOXC1 promotes proliferation and epithelial-mesenchymal transition in cervical carcinoma through the PI3K-AKT signal pathway. Am. J. Transl. Res. 2017, 9, 1297–1306. [Google Scholar] [PubMed]
- Wang, L.; Chai, L.; Ji, Q.; Cheng, R.; Wang, J.; Han, S. Forkhead box protein C1 promotes cell proliferation and invasion in human cervical cancer. Mol. Med. Rep. 2018, 17, 4392–4398. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Tian, J.; Hao, Q.; Yin, L.R. MicroRNA-495 downregulates FOXC1 expression to suppress cell growth and migration in endometrial cancer. Tumor Biol. 2016, 37, 239–251. [Google Scholar] [CrossRef]
- Chung, T.K.; Lau, T.S.; Cheung, T.H.; Yim, S.F.; Lo, K.W.; Siu, N.S.; Chan, L.K.; Yu, M.Y.; Kwong, J.; Doran, G.; et al. Dysregulation of microRNA-204 mediates migration and invasion of endometrial cancer by regulating FOXC1. Int. J. Cancer 2012, 130, 1036–1045. [Google Scholar] [CrossRef]
- Xu, Y.; Shao, Q.; Yao, H.; Jin, Y.; Ma, Y.; Jia, L. Overexpression of FOXC1 correlates with poor prognosis in gastric cancer patients. Histopathology 2014, 64, 963–970. [Google Scholar] [CrossRef]
- Gao, W.; Wu, Y.; He, X.; Zhang, C.; Zhu, M.; Chen, B.; Liu, Q.; Qu, X.; Li, W.; Wen, S.; et al. MicroRNA-204-5p inhibits invasion and metastasis of laryngeal squamous cell carcinoma by suppressing forkhead box C1. J. Cancer 2017, 8, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, L.; Xiao, S.J.; Liu, P.; Yi, S.J.; Zhang, X.L.; Ou-Yang, S.; Tan, S.K.; Lei, X. Forkhead box C1 induces epithelial-mesenchymal transition and is a potential therapeutic target in nasopharyngeal carcinoma. Mol. Med. Rep. 2015, 12, 8003–8009. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Huang, W.; Tian, D.; Zhu, H.; Qi, X.; Chen, Z.; Zhang, Y.; Hu, H.; Fan, D.; Nie, Y.; et al. Overexpression of forkhead box C1 promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Hepatology 2013, 57, 610–624. [Google Scholar] [CrossRef]
- Wei, L.X.; Zhou, R.S.; Xu, H.F.; Wang, J.Y.; Yuan, M.H. High expression of FOXC1 is associated with poor clinical outcome in non-small cell lung cancer patients. Tumour Biol. 2013, 34, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Wang, Z.; Gao, X.; He, W.; Cai, Y.; Chen, H.; Xu, R. FOXC1 induces cancer stem cell-like properties through upregulation of beta-catenin in NSCLC. J. Exp. Clin. Cancer Res. 2018, 37, 220. [Google Scholar] [CrossRef] [PubMed]
- Blonska, M.; Zhu, Y.; Chuang, H.H.; You, M.J.; Kunkalla, K.; Vega, F.; Lin, X. Jun-regulated genes promote interaction of diffuse large B-cell lymphoma with the microenvironment. Blood 2015, 125, 981–991. [Google Scholar] [CrossRef]
- Steidl, C.; Diepstra, A.; Lee, T.; Chan, F.C.; Farinha, P.; Tan, K.; Telenius, A.; Barclay, L.; Shah, S.P.; Connors, J.M.; et al. Gene expression profiling of microdissected Hodgkin Reed-Sternberg cells correlates with treatment outcome in classical Hodgkin lymphoma. Blood 2012, 120, 3530–3540. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Liu, S.; Zhao, Y.; Wang, L.; Du, G. FOXC1 promotes melanoma by activating MST1R/PI3K/AKT pathway and is associated with poor prognosis in melanoma. Oncotarget 2016, 7, 84375–84387. [Google Scholar] [CrossRef]
- Wang, L.; Gu, F.; Liu, C.Y.; Wang, R.J.; Li, J.; Xu, J.Y. High level of FOXC1 expression is associated with poor prognosis in pancreatic ductal adenocarcinoma. Tumour Biol. 2013, 34, 853–858. [Google Scholar] [CrossRef]
- Subramani, R.; Camacho, F.A.; Levin, C.I.; Flores, K.; Clift, A.; Galvez, A.; Terres, M.; Rivera, S.; Kolli, S.N.; Dodderer, J.; et al. FOXC1 plays a crucial role in the growth of pancreatic cancer. Oncogenesis 2018, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Yao, J.; Chen, Y.; Zhou, C.; Geng, P.; Mao, H.; Fang, X. A novel long non-coding RNA FOXCUT and mRNA FOXC1 pair promote progression and predict poor prognosis in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 2838–2849. [Google Scholar] [PubMed]
- Zhu, X.; Wei, L.; Bai, Y.; Wu, S.; Han, S. FoxC1 promotes epithelial-mesenchymal transition through PBX1 dependent transactivation of ZEB2 in esophageal cancer. Am. J. Cancer Res. 2017, 7, 1642–1653. [Google Scholar] [PubMed]
- Deng, L.; Liu, T.; Zhang, B.; Wu, H.; Zhao, J.; Chen, J. Forkhead box C1 is targeted by microRNA-133b and promotes cell proliferation and migration in osteosarcoma. Exp. Ther. Med. 2017, 14, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Li, L.S.; Yang, Z. Correlation of FOXC1 protein with clinicopathological features in serous ovarian tumors. Oncol. Lett. 2016, 11, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Assi, S.A.; Imperato, M.R.; Coleman, D.J.L.; Pickin, A.; Potluri, S.; Ptasinska, A.; Chin, P.S.; Blair, H.; Cauchy, P.; James, S.R.; et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat. Genet. 2018, 51, 151–162. [Google Scholar] [CrossRef]
- Cauchy, P.; James, S.R.; Zacarias-Cabeza, J.; Ptasinska, A.; Imperato, M.R.; Assi, S.A.; Piper, J.; Canestraro, M.; Hoogenkamp, M.; Raghavan, M.; et al. Chronic FLT3-ITD Signaling in Acute Myeloid Leukemia Is Connected to a Specific Chromatin Signature. Cell Rep. 2015, 12, 821–836. [Google Scholar] [CrossRef]
- Jin, Y.; Han, B.; Chen, J.; Wiedemeyer, R.; Orsulic, S.; Bose, S.; Zhang, X.; Karlan, B.Y.; Giuliano, A.E.; Cui, Y.; et al. FOXC1 is a critical mediator of EGFR function in human basal-like breast cancer. Ann. Surg. Oncol. 2014, 21, 758–766. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jiao, S.; Jia, Y.; Li, Y. Effects of targeted silencing of FOXC1 gene on proliferation and in vitro migration of human non-small-cell lung carcinoma cells. Am. J. Transl. Res. 2016, 8, 3309–3318. [Google Scholar] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Zhou, B.; Qu, Y.; Gao, B.; Xu, Y.; Chung, S.; Yang, S.X.; Ivy, S.P. FOXC1-induced non-canonical WNT5A-MMP7 signaling regulates invasiveness in triple-negative breast cancer. Oncogene 2018, 37, 1399. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Balczerski, B.; Ciozda, A.; Louie, K.; Oralova, V.; Huysseune, A.; Crump, J.G. Fox proteins are modular competency factors for facial cartilage and tooth specification. Development 2018, 145, dev165498. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Shyu, W.C.; Chang, C.W.; Wang, C.C.; Wu, C.P.; Lee, H.T.; Chen, L.J.; Hsieh, C.H. Tumor Hypoxia Regulates Forkhead Box C1 to Promote Lung Cancer Progression. Theranostics 2017, 7, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, Z.; Zhang, L.; Tian, D.; Wang, D.; Fan, D.; Wu, K.; Xia, L. Interleukin-8 Induces Expression of FOXC1 to Promote Transactivation of CXCR1 and CCL2 in Hepatocellular Carcinoma Cell Lines and Formation of Metastases in Mice. Gastroenterology 2015, 149, 1053–1067. [Google Scholar] [CrossRef]
- Seo, S.; Singh, H.P.; Lacal, P.M.; Sasman, A.; Fatima, A.; Liu, T.; Schultz, K.M.; Losordo, D.W.; Lehmann, O.J.; Kume, T. Forkhead box transcription factor FoxC1 preserves corneal transparency by regulating vascular growth. Proc. Natl. Acad. Sci. USA 2012, 109, 2015–2020. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Gabbasov, R.; Xiao, F.; Howe, C.G.; Bickel, L.E.; O’Brien, S.W.; Benrubi, D.; Do, T.V.; Zhou, Y.; Nicolas, E.; Cai, K.Q.; et al. NEDD9 promotes oncogenic signaling, a stem/mesenchymal gene signature, and aggressive ovarian cancer growth in mice. Oncogene 2018, 37, 4854–4870. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, S.T.; Keri, R.A. The Forkhead Box Transcription Factor FOXC1 Promotes Breast Cancer Invasion by Inducing Matrix Metalloprotease 7 (MMP7) Expression. J. Biol. Chem. 2012, 287, 24631–24640. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Y.; Ding, S.M.; Zhou, L.; Xie, H.Y.; Chen, K.J.; Zhang, W.; Xing, C.Y.; Guo, H.J.; Zheng, S.S. FOXC1 Contributes to Microvascular Invasion in Primary Hepatocellular Carcinoma Via Regulating Epithelial-Mesenchymal Transition. Int. J. Biol. Sci. 2012, 8, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Choi, P.S.; Chaffer, C.L.; Labella, K.; Hwang, J.H.; Giacomelli, A.O.; Kim, J.W.; Ilic, N.; Doench, J.G.; Ly, S.H.; et al. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife 2018, 7, e37184. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Li, L.; Ou, Z.; Kong, C.; Zhang, Y.; Dong, Z.; Zhu, S.; Jiang, H.; Shao, Z.; Huang, B.; et al. FOXC1, a target of polycomb, inhibits metastasis of breast cancer cells. Breast Cancer Res. Treat. 2012, 131, 65–73. [Google Scholar] [CrossRef]
- Tkocz, D.; Crawford, N.T.; Buckley, N.E.; Berry, F.B.; Kennedy, R.D.; Gorski, J.J.; Harkin, D.P.; Mullan, P.B. BRCA1 and GATA3 corepress FOXC1 to inhibit the pathogenesis of basal-like breast cancers. Oncogene 2012, 31, 3667–3678. [Google Scholar] [CrossRef]
- Prat, A.; Cruz, C.; Hoadley, K.A.; Díez, O.; Perou, C.M.; Balmaña, J. Molecular features of the basal-like breast cancer subtype based on BRCA1 mutation status. Breast Cancer Res. Treat. 2014, 147, 185–191. [Google Scholar] [CrossRef]
- Dejeux, E.; Rønneberg, J.A.; Solvang, H.; Bukholm, I.; Geisler, S.; Aas, T.; Gut, I.G.; Børresen-Dale, A.L.; Lønning, P.E.; Kristensen, V.N.; et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol. Cancer 2010, 9, 68. [Google Scholar] [CrossRef]
- Muggerud, A.A.; Rønneberg, J.A.; Wärnberg, F.; Botling, J.; Busato, F.; Jovanovic, J.; Solvang, H.; Bukholm, I.; Børresen-Dale, A.L.; Kristensen, V.N.; et al. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res. 2010, 12, R3. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Jin, Y.; Han, B.; Qu, Y.; Gao, B.; Giuliano, A.E.; Cui, X. Identification of EGF-NF-κB-FOXC1 signaling axis in basal-like breast cancer. Cell Commun. Signal. 2017, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ray, P.S.; Sim, M.S.; Zhou, X.Z.; Lu, K.P.; Lee, A.V.; Lin, X.; Bagaria, S.P.; Giuliano, A.E.; Cui, X. FOXC1 regulates the functions of human basal-like breast cancer cells by activating NF-κB signaling. Oncogene 2012, 31, 4798–4802. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kume, T. Foxc Transcription Factors Directly Regulate Dll4 and Hey2 Expression by Interacting with the VEGF-Notch Signaling Pathways in Endothelial Cells. PLoS ONE 2008, 3, e2401. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Katsuno, Y.; Ehata, S.; Miyazono, K. Autocrine TGF-β protects breast cancer cells from apoptosis through reduction of BH3-only protein. Bim. J. Biochem. 2011, 149, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Jiang, S.; Cheng, Y.; Li, T.; Hu, W.; Ma, Z.; Chen, F.; Yang, Y. FOXC1 in cancer development and therapy: Deciphering its emerging and divergent roles. Ther. Adv. Med. Oncol. 2017, 9, 797–816. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yao, J.; Luo, W.; Feng, F.; Ma, J.; Ren, Y.; Wang, D.L.; Bu, R.F. The expression and functional role of a FOXC1 related mRNA-lncRNA pair in oral squamous cell carcinoma. Mol. Cell. Biochem. 2014, 394, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shen, L.; Yao, J.; Li, Y.; Wang, Y.; Chen, H.; Geng, P. Forkhead box C1 promoter upstream transcript, a novel long non-coding RNA, regulates proliferation and migration in basal-like breast cancer. Mol. Med. Rep. 2015, 11, 3155–3159. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Post-transcriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef]
- Jensen, T.W.; Ray, T.; Wang, J.; Li, X.; Naritoku, W.Y.; Han, B.; Bellafiore, F.; Bagaria, S.P.; Qu, A.; Cui, X.; et al. Diagnosis of Basal-Like Breast Cancer Using a FOXC1-Based Assay. J. Natl. Cancer Inst. 2015, 107, djv148. [Google Scholar] [CrossRef]
- Han, B.; Bhowmick, N.; Qu, Y.; Chung, S.; Giuliano, A.E.; Cui, X. FOXC1: An emerging marker and therapeutic target for cancer. Oncogene 2017, 36, 3957–3963. [Google Scholar] [CrossRef] [PubMed]
- Yu-Rice, Y.; Jin, Y.; Han, B.; Qu, Y.; Johnson, J.; Watanabe, T.; Cheng, L.; Deng, N.; Tanaka, H.; Gao, B.; et al. FOXC1 is involved in ERα silencing by counteracting GATA3 binding and is implicated in endocrine resistance. Oncogene 2016, 35, 5400–5411. [Google Scholar] [CrossRef] [PubMed]
- Kume, T.; Shackour, T. Meta-analysis of the likelihood of FOXC1 expression in early- and late-stage tumors. Oncotarget 2018, 9, 36625–36630. [Google Scholar] [CrossRef] [PubMed]
- Swinstead, E.E.; Miranda, T.B.; Paakinaho, V.; Baek, S.; Goldstein, I.; Hawkins, M.; Karpova, T.S.; Ball, D.; Mazza, D.; Lavis, L.D.; et al. Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell 2016, 165, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Zhou, X.; Liu, X.; Wang, Y.; Su, D.; Hou, Y.; Yu, N.; Yang, C.; Liu, B.; Gao, J.; et al. FOXK2 Elicits Massive Transcription Repression and Suppresses the Hypoxic Response and Breast Cancer Carcinogenesis. Cancer Cell 2016, 30, 708–722. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.R.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.K.; Ray, T.; Ostadhossein, F.; Kim, B.; Ray, P.S.; Pan, D. Carotenoid Nanovector for Efficient Therapeutic Gene Knockdown of Transcription Factor FOXC1 in Liver Cancer. Bioconjug. Chem. 2016, 27, 594–603. [Google Scholar] [CrossRef] [PubMed]
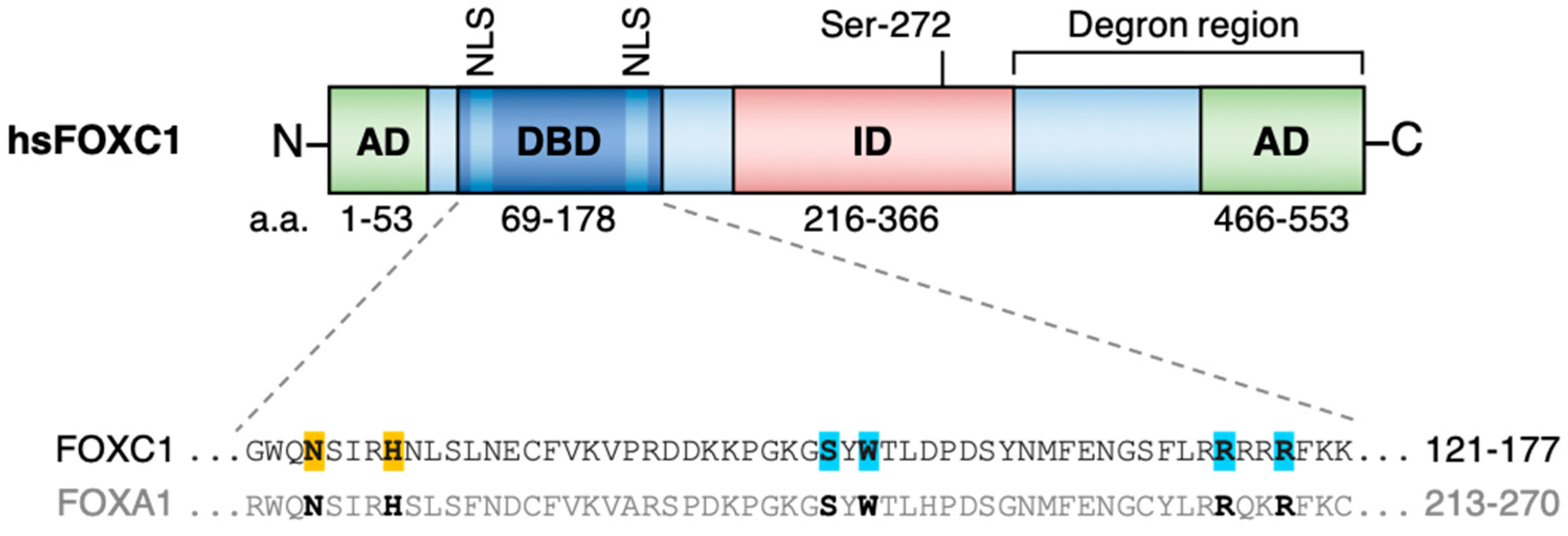

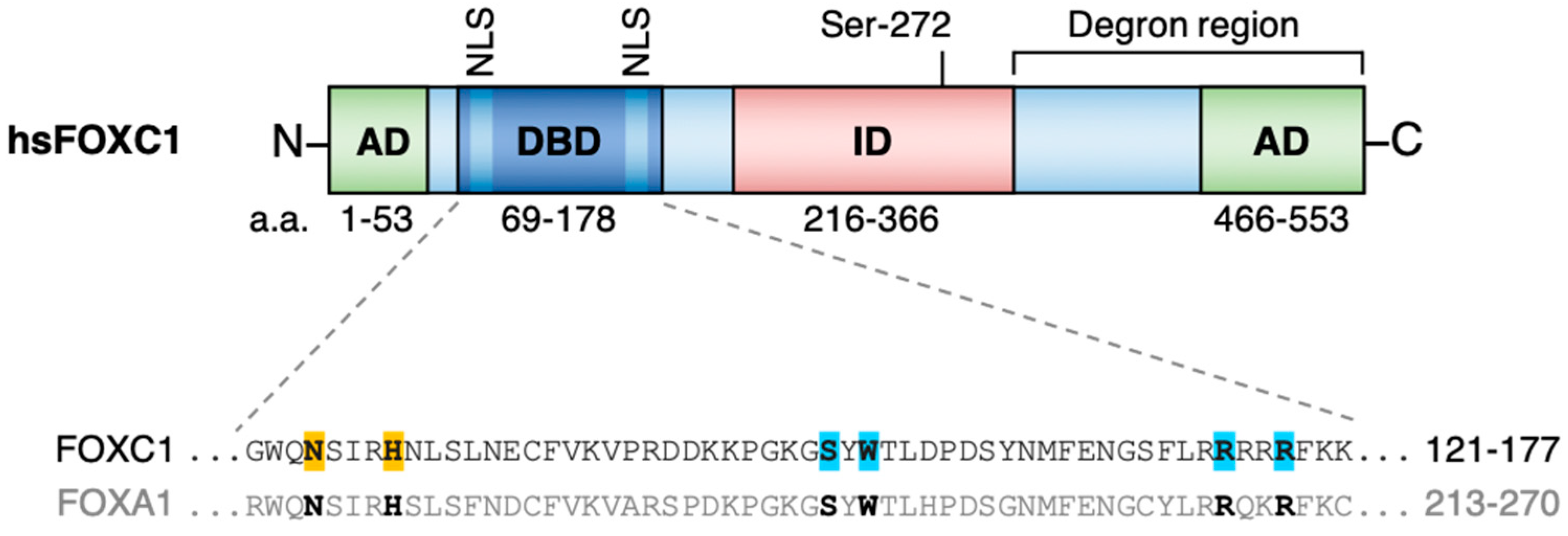{kind=link}
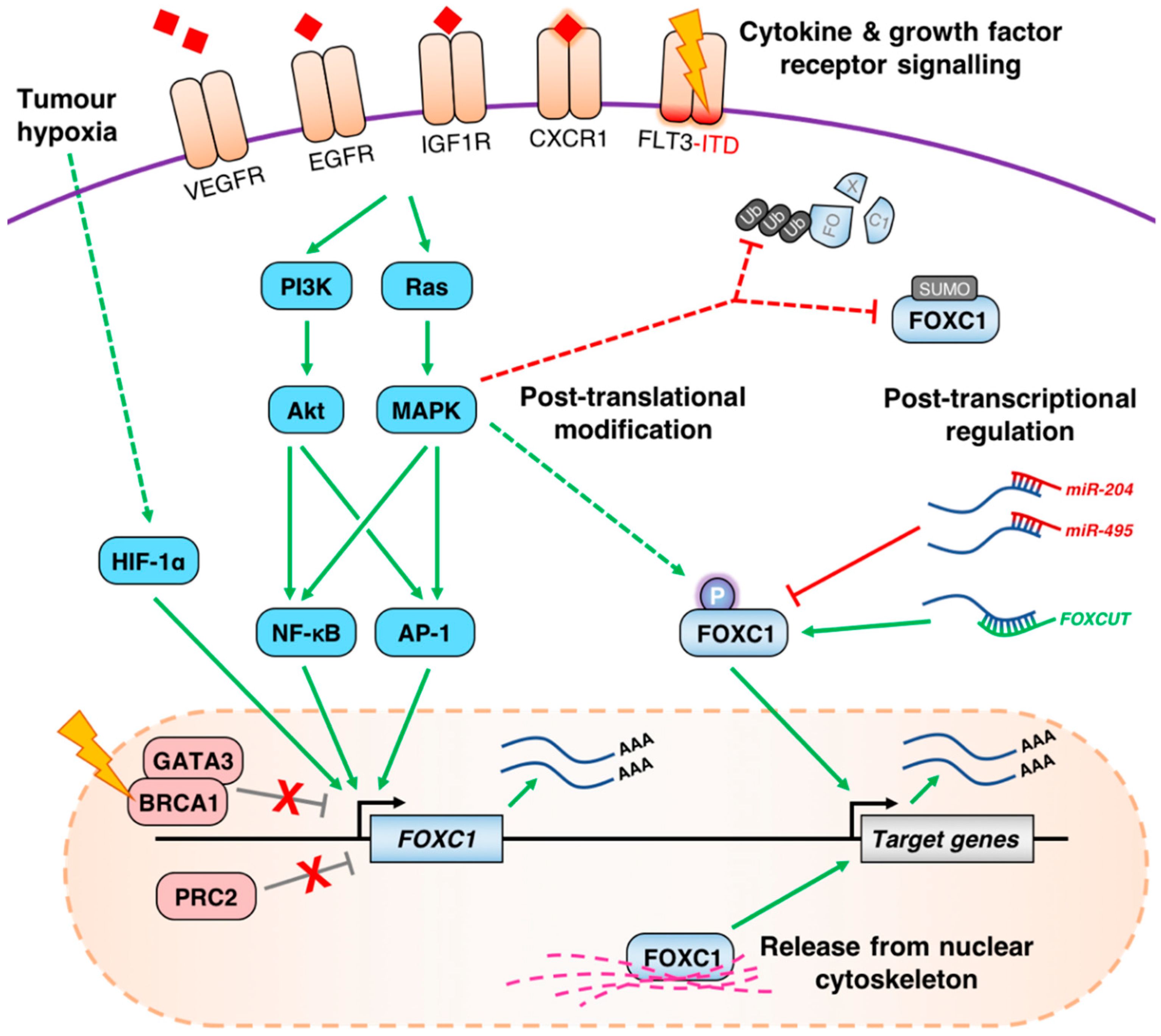{kind=link}
| Cancer Type | FOXC1 Alteration | Associated Clinical Outcome | Case Sizes | Functional Impact of Experimental FOXC1 Manipulation | |
|---|---|---|---|---|---|
| Overexpression | Silencing | ||||
| AML | Overexpression | Inferior OS [40,41]; increased risk of relapse and resistance to induction chemotherapy [41] | 270 [40] 765 [41] | Co-operates in vivo with Hoxa9 to block differentiation and accelerate onset of AML [40] | Loss of clonogenic potential and enhanced morphological differentiation in vitro [40] |
| Breast (BLBC and Luminal B) | Overexpression | BLBC: Increased metastasis; inferior OS [42] | 2073 | BLBC: Increased cell growth/survival [42], induced progenitor-like phenotype in vitro [35]; enhanced CSC-like properties in vitro and in vivo [44]. | BLBC: Impaired cell proliferation, migration and invasion in vitro [42]. |
| Luminal B: improved RFS [43] | 1142 | Luminal B: impaired invasion and lung metastasis formation [43]. | - | ||
| Colorectal | Overexpression | Increased risk of metastasis; inferior OS/RFS [45,46] | 361 [45] 120 [46] | Increased tumorigenicity and metastasis in vivo [45,46]; enhanced Warburg effect in vitro [45]. | Impaired tumorigenicity and metastasis in vivo [45,46]; inhibited Warburg effect in vitro [45]. |
| Cervical | Overexpression | Increased risk of invasion, metastasis and relapse; inferior OS [47] | 336 | - | Suppressed cell growth, migration and invasion in vitro [48]. |
| Endometrial | Overexpression | - | 20 | Enhanced migration and colony formation in vitro [49,50]. | Increased apoptosis in vitro, impaired tumor growth in vivo [49]. |
| Gastric | Overexpression | Inferior OS/RFS [51] | 120 | - | - |
| Head and Neck (LSCC and NPC) | Overexpression | LSCC: increased risk of lymph node metastasis [52] | 147 | - | - |
| NPC: increased risk of metastasis [53] | 93 | - | - | ||
| Liver (HCC) | Overexpression | Inferior OS; increased relapse risk [54] | 406 | Increased cell invasion in vitro and metastasis in vivo [54]. | Impaired cell invasion in vitro and metastasis in vivo [54]. |
| Lung (NSCLC) | Overexpression | Inferior OS/RFS [55,56] | 125 [55] 1129 [56] | Enhanced CSC-like properties, drug resistance and tumorigenicity in vivo [56]. | Suppressed self-renewal and impaired tumorigenicity in vivo [56]. |
| Lymphoma (DLBCL and HL) | Overexpression | DLBCL: trend towards increased extranodal spread [57] | 25 | - | - |
| HL: part of Hodgkin Reed-Sternberg cell-specific gene signature correlated with treatment failure [58] | 29 | - | - | ||
| Melanoma | Overexpression | Inferior OS; increased risk of metastasis [59] | 228 | Enhanced cell proliferation and invasion in vitro [59] | - |
| Pancreatic (PDA) | Overexpression | Increased risk of lymph node metastasis, inferior OS [60] | 30 | Increased cell proliferation and invasion in vitro; promoted tumorigenicity in vivo [61] | Impaired cell proliferation and invasion in vitro [61] |
| Esophageal (ESCC) | Overexpression | ESCC: increased rate of metastasis, inferior OS [62]; | 84 | ESCC: promoted ESCC cell proliferation, colony formation and invasion in vitro [63] | ESCC: impaired ESCC cell colony formation, and invasion in vitro [62,63] |
| Osteosarcoma | Overexpression | Higher TNM staging [64] | 42 | - | Impaired cell proliferation and migration in vitro [64] |
| Ovarian (Serous) | Overexpression | Improved OS [65] | 80 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilding, L.N.; Somervaille, T.C.P. The Diverse Consequences of FOXC1 Deregulation in Cancer. Cancers 2019, 11, 184. https://doi.org/10.3390/cancers11020184
Gilding LN, Somervaille TCP. The Diverse Consequences of FOXC1 Deregulation in Cancer. Cancers. 2019; 11(2):184. https://doi.org/10.3390/cancers11020184
Chicago/Turabian StyleGilding, L. Niall, and Tim C. P. Somervaille. 2019. "The Diverse Consequences of FOXC1 Deregulation in Cancer" Cancers 11, no. 2: 184. https://doi.org/10.3390/cancers11020184
APA StyleGilding, L. N., & Somervaille, T. C. P. (2019). The Diverse Consequences of FOXC1 Deregulation in Cancer. Cancers, 11(2), 184. https://doi.org/10.3390/cancers11020184