High-Throughput Screening Identified Compounds Sensitizing Tumor Cells to Glucose Starvation in Culture and VEGF Inhibitors In Vivo

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cell Viability Assays
2.3. Cell Death Assays
2.4. Cell Lysis and Sample Preparation
2.5. Gel Electrophoresis and Immunoblotting
2.6. Antibodies
2.7. Tumor Xenografts and Drug Treatments in Vivo
2.8. Immunohistochemistry (IHC) and Analysis
2.9. Statistics
3. Results
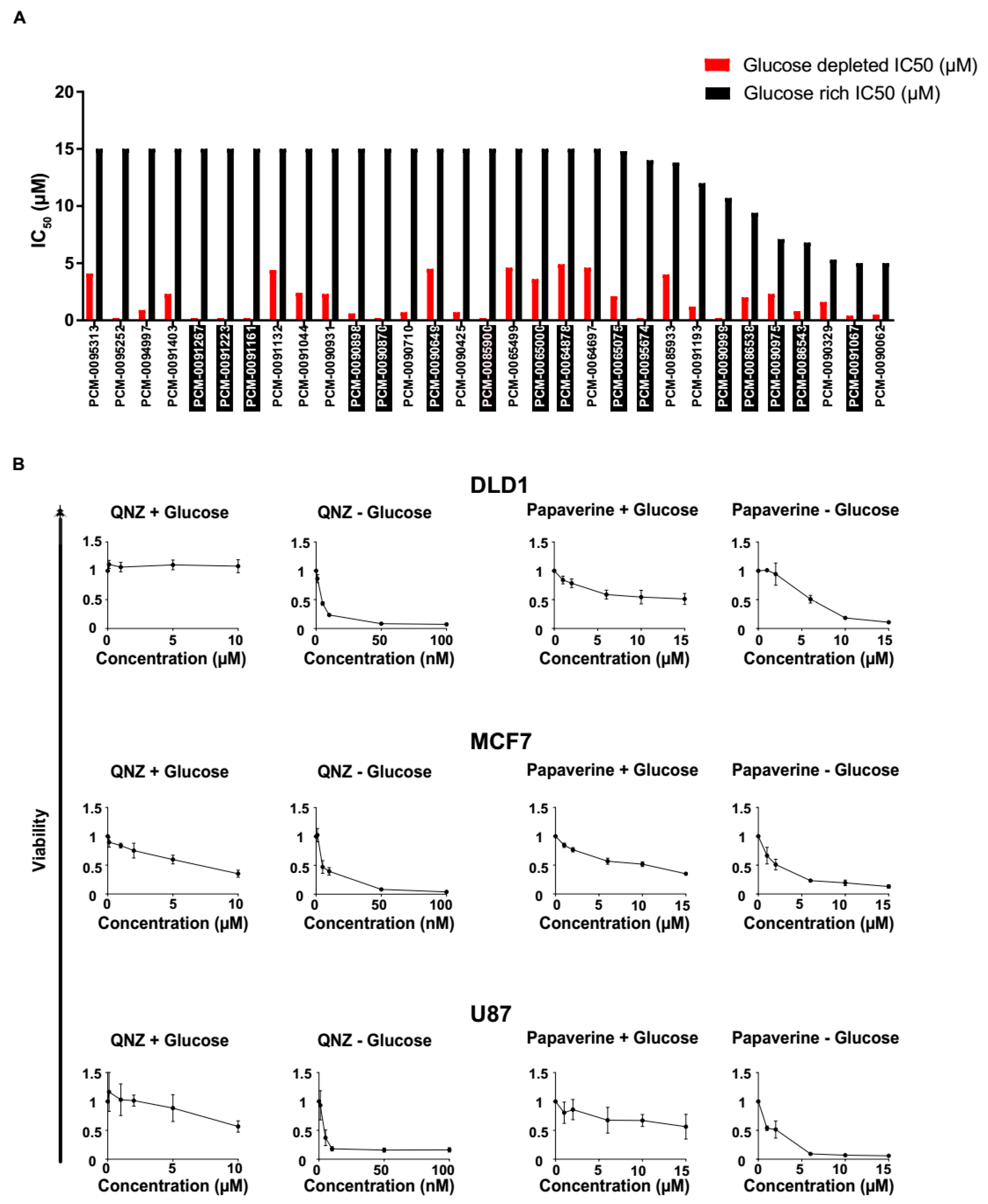
3.1. High-Throughput Synthetic Lethality Drug Screening for Selectively Potent Compounds under Glucose Starvation
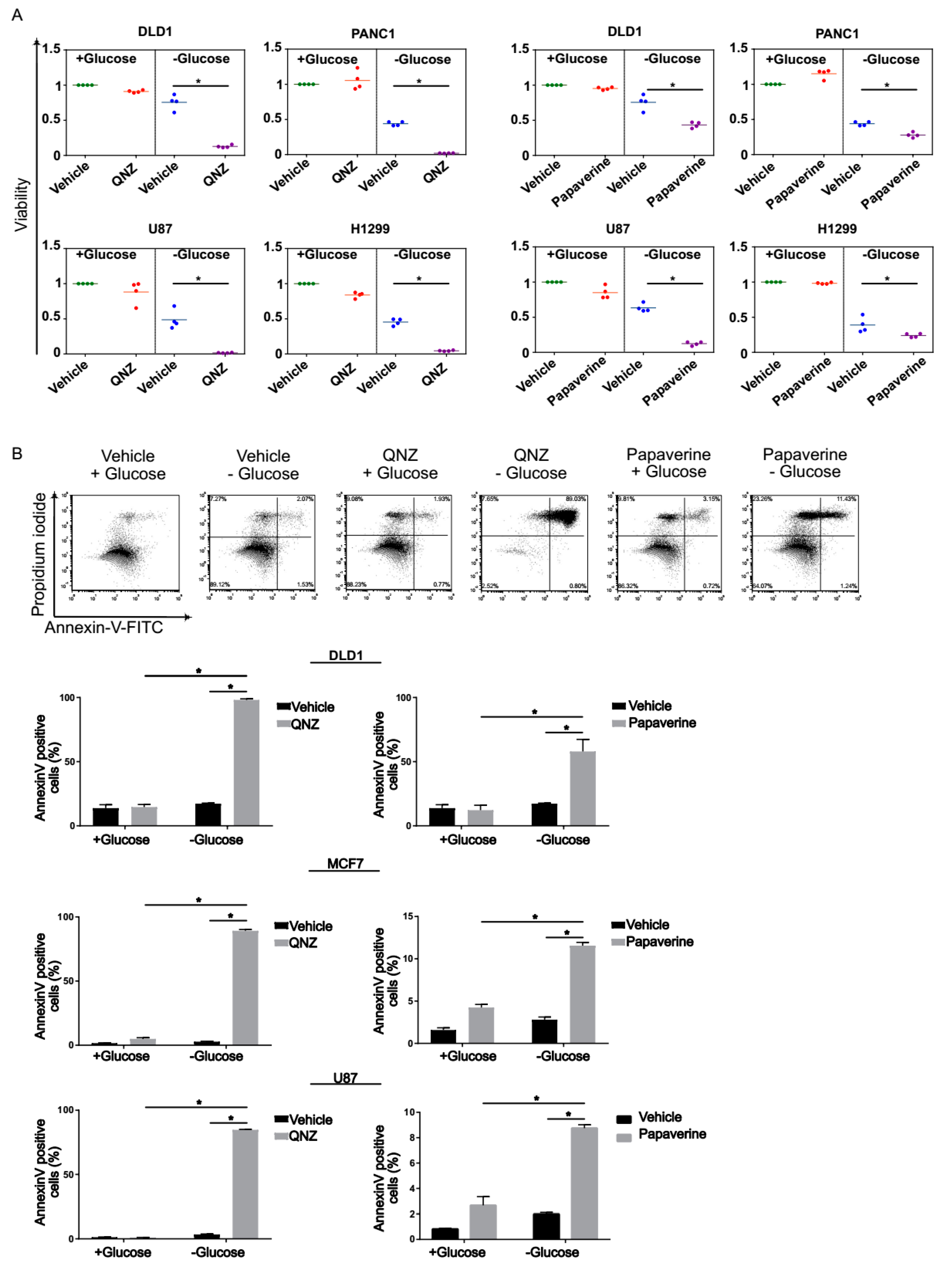
3.2. Screen Validation and Identification of QNZ and Papaverine as Compounds with Selective Toxicity under Glucose Starvation
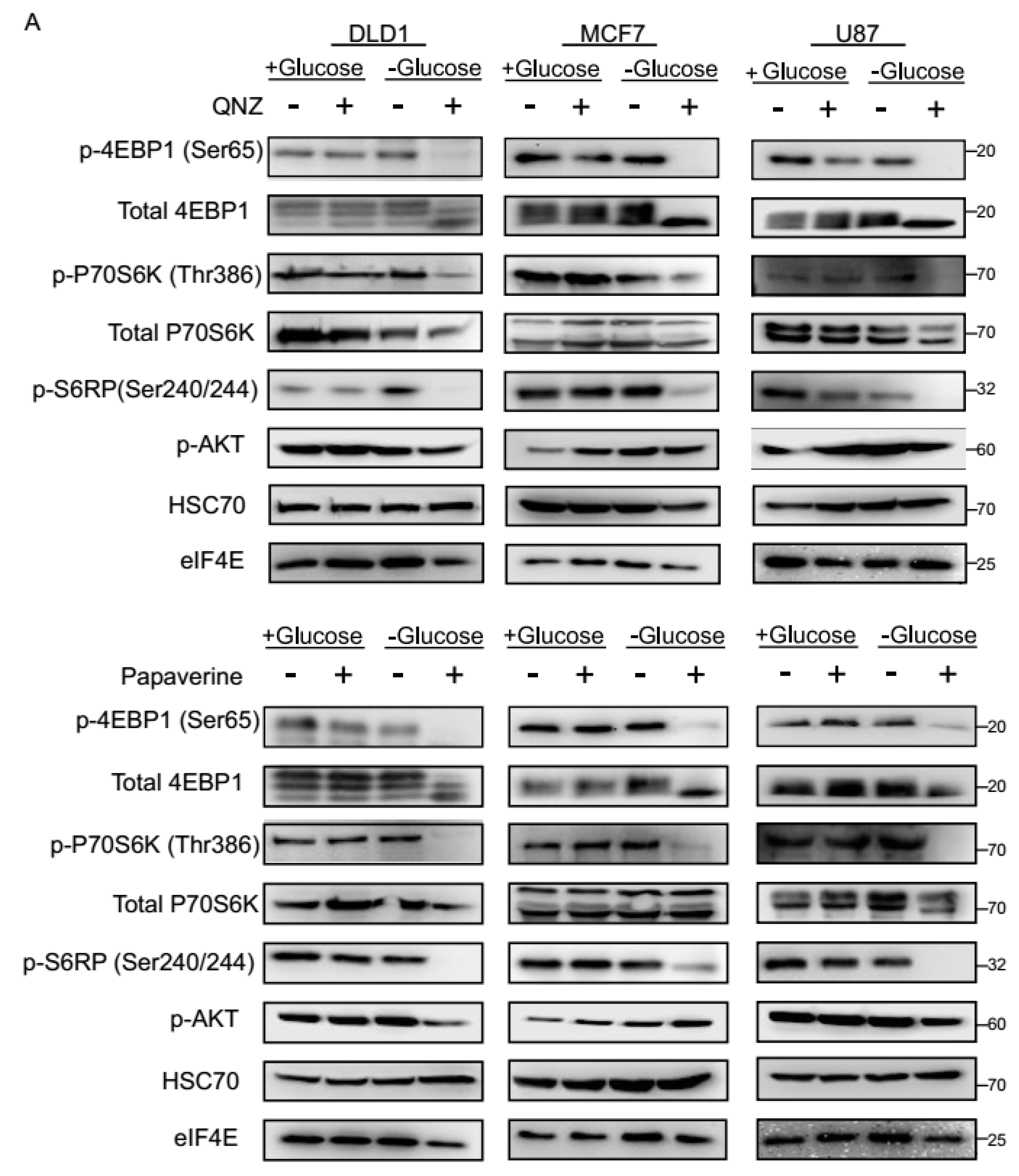
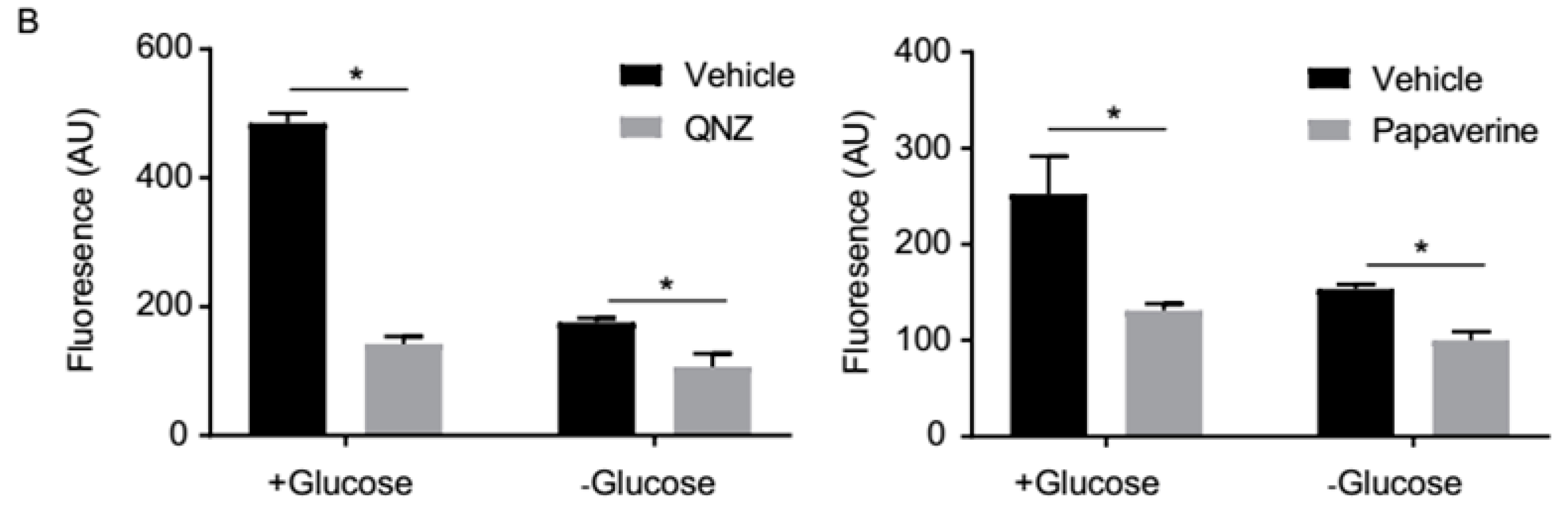
3.3. QNZ and Papaverine Inhibit the mTOR Pathway Selectively under Glucose Starvation
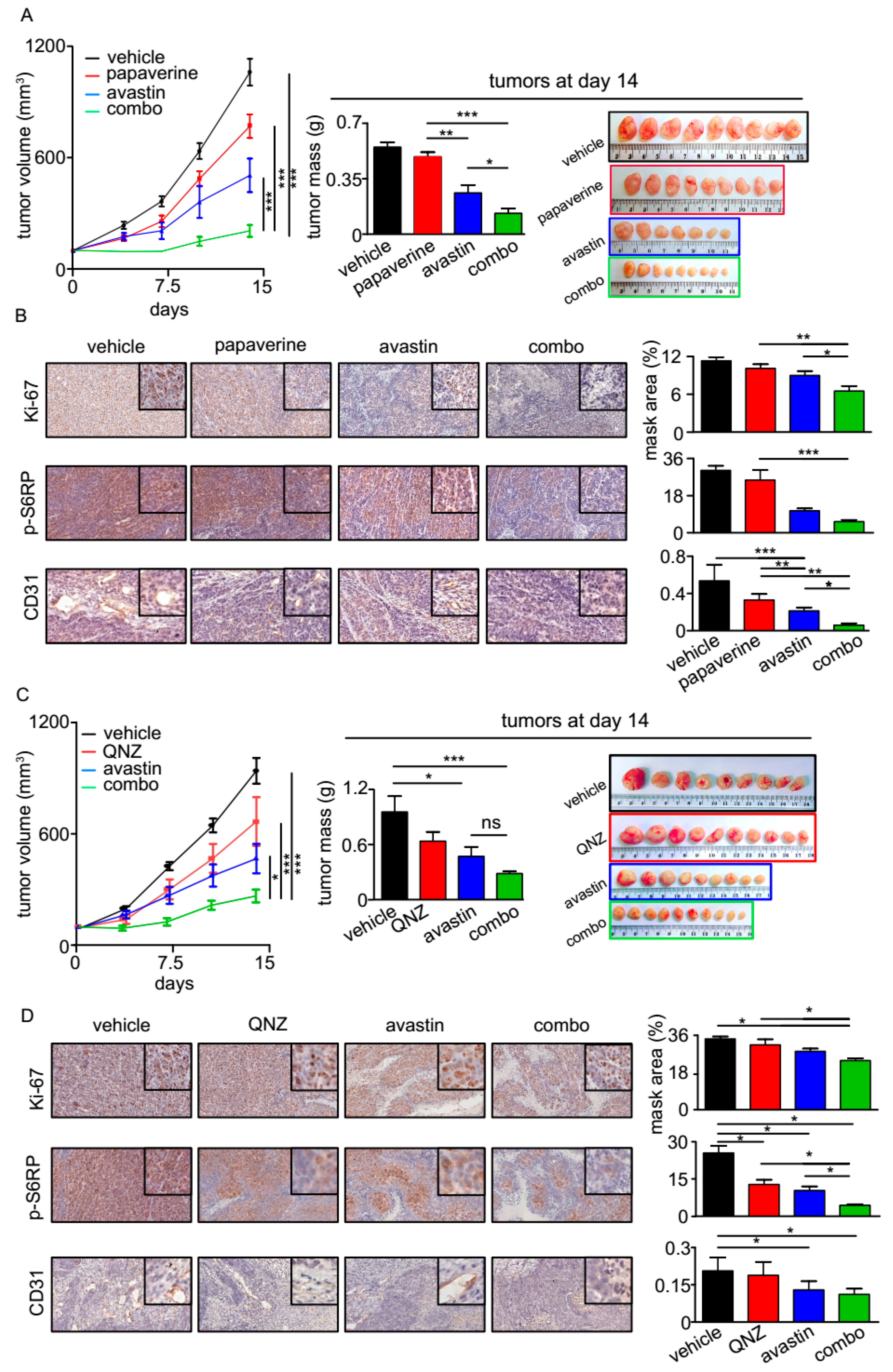
3.4. QNZ and Papaverine Enhance the Antitumor Activity of Bevacizumab in Vivo
4. Discussion
Limitations of Our Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hay, N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, H.G. The carbohydrate metabolism of certain pathological overgrowths. Biochem. J. 1928, 22, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Gao, M.; Liang, J.; Lu, Y.; Guo, H.; German, P.; Bai, S.; Jonasch, E.; Yang, X.; Mills, G.B.; Ding, Z. Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene 2014, 33, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Komurov, K.; Tseng, J.-T.; Muller, M.; Seviour, E.G.; Moss, T.J.; Yang, L.; Nagrath, D.; Ram, P.T. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol. Syst. Biol. 2012, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. MTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.-R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Kim, S.G.; Vander Heiden, M.G.; Mahoney, S.J.; Vu, H.; Yoon, S.O.; Cantley, L.C.; Blenis, J. Glucose Addiction of TSC Null Cells Is Caused by Failed mTORC1-Dependent Balancing of Metabolic Demand with Supply. Mol. Cell 2010, 38, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. MTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-KB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Publ. Gr. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Krishnathas, R.; Bonke, E.; Dröse, S.; Zickermann, V.; Nasiri, H.R. Identification of 4-N-[2-(4-phenoxyphenyl)ethyl]quinazoline-4,6-diamine as a novel, highly potent and specific inhibitor of mitochondrial complex I. Medchemcomm 2017, 8, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, N.; Nakagawa-Hattori, Y.; Mizuno, Y. Effect of dopamine, dimethoxyphenylethylamine, papaverine, and related compounds on mitochondrial respiration and complex I activity. J. Neurochem. 1996, 66, 1174–1181. [Google Scholar] [CrossRef]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.-R.F.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef] [PubMed]
- Musa, J.; Orth, M.F.; Dallmayer, M.; Baldauf, M.; Pardo, C.; Rotblat, B.; Kirchner, T.; Leprivier, G.; Grünewald, T.G.P. Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): A master regulator of mRNA translation involved in tumorigenesis. Oncogene 2016, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Li, C.; Xing, Z.; Hu, Q.; Liang, K.; Han, L.; Wang, C.; Hawke, D.H.; Wang, S.; Zhang, Y.; et al. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat. Cell Biol. 2016, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rotblat, B.; Grunewald, T.G.P.; Leprivier, G.; Melino, G.; Knight, R.A. Anti-oxidative stress response genes: bioinformatic analysis of their expression and relevance in multiple cancers. Oncotarget 2013, 4, 2577–2590. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro. Oncol. 2017, 1, 43–54. [Google Scholar] [CrossRef]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.-T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef]
- Cetinbas, N.; Daugaard, M.; Mullen, A.R.R.; Hajee, S.; Rotblat, B.; Lopez, A.; Li, A.; DeBerardinis, R.J.J.; Sorensen, P.H.H. Loss of the tumor suppressor Hace1 leads to ROS-dependent glutamine addiction. Oncogene 2014, 34, 1–6. [Google Scholar] [CrossRef]
- Guan, B.J.; van Hoef, V.; Jobava, R.; Elroy-Stein, O.; Valasek, L.S.; Cargnello, M.; Gao, X.H.; Krokowski, D.; Merrick, W.C.; Kimball, S.R.; et al. A Unique ISR Program Determines Cellular Responses to Chronic Stress. Mol. Cell 2017, 68, 885–900.e6. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim-Hashim, A.; Robertson-Tessi, M.; Enriquez-Navas, P.M.; Damaghi, M.; Balagurunathan, Y.; Wojtkowiak, J.W.; Russell, S.; Yoonseok, K.; Lloyd, M.C.; Bui, M.M.; et al. Defining cancer subpopulations by adaptive strategies rather than molecular properties provides novel insights into intratumoral evolution. Cancer Res. 2017, 77, 2242–2254. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 1–12. [Google Scholar] [CrossRef]
- Mai, W.X.; Gosa, L.; Daniels, V.W.; Ta, L.; Tsang, J.E.; Higgins, B.; Gilmore, W.B.; Bayley, N.A.; Harati, M.D.; Lee, J.T.; et al. Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat. Med. 2017, 23, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef] [PubMed]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G.; Andrzejewski, S.; et al. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.L.; Leprivier, G.; Robertson, M.D.; Chow, C.; Martin, M.J.; Laderoute, K.R.; Davicioni, E.; Triche, T.J.; Sorensen, P.H.B. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012, 19, 501–510. [Google Scholar] [CrossRef]
- Leprivier, G.; Rotblat, B.; Khan, D.; Jan, E.; Sorensen, P.H. Stress-mediated translational control in cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1849, 845–860. [Google Scholar] [CrossRef]
- Robichaud, N.; Sonenberg, N. Translational control and the cancer cell response to stress. Curr. Opin. Cell Biol. 2017, 45, 102–109. [Google Scholar] [CrossRef]
- Ilagan, E.; Manning, B.D. Emerging Role of mTOR in the Response to Cancer Therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.-Y.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Kassell, N.F.; Helm, G.; Simmons, N.; Phillips, C.D.; Cail, W.S. Treatment of cerebral vasospasm with intra-arterial papaverine. J. Neurosurg. 1992, 77, 848–852. [Google Scholar] [CrossRef]
- Virag, R. Intracavernous injection of papaverine for erectile failure. J. Urol. 2002, 167, 1196. [Google Scholar] [CrossRef]
- Benej, M.; Hong, X.; Vibhute, S.; Scott, S.; Wu, J.; Graves, E.; Le, Q.-T.; Koong, A.C.; Giaccia, A.J.; Yu, B.; et al. Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 10756–10761. [Google Scholar] [CrossRef]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marciano, R.; Prasad, M.; Ievy, T.; Tzadok, S.; Leprivier, G.; Elkabets, M.; Rotblat, B. High-Throughput Screening Identified Compounds Sensitizing Tumor Cells to Glucose Starvation in Culture and VEGF Inhibitors In Vivo. Cancers 2019, 11, 156. https://doi.org/10.3390/cancers11020156
Marciano R, Prasad M, Ievy T, Tzadok S, Leprivier G, Elkabets M, Rotblat B. High-Throughput Screening Identified Compounds Sensitizing Tumor Cells to Glucose Starvation in Culture and VEGF Inhibitors In Vivo. Cancers. 2019; 11(2):156. https://doi.org/10.3390/cancers11020156
Chicago/Turabian StyleMarciano, Ran, Manu Prasad, Tal Ievy, Sapir Tzadok, Gabriel Leprivier, Moshe Elkabets, and Barak Rotblat. 2019. "High-Throughput Screening Identified Compounds Sensitizing Tumor Cells to Glucose Starvation in Culture and VEGF Inhibitors In Vivo" Cancers 11, no. 2: 156. https://doi.org/10.3390/cancers11020156
APA StyleMarciano, R., Prasad, M., Ievy, T., Tzadok, S., Leprivier, G., Elkabets, M., & Rotblat, B. (2019). High-Throughput Screening Identified Compounds Sensitizing Tumor Cells to Glucose Starvation in Culture and VEGF Inhibitors In Vivo. Cancers, 11(2), 156. https://doi.org/10.3390/cancers11020156