Companion Animals as Models for Inhibition of STAT3 and STAT5

 ,
,
Abstract
1. Introduction
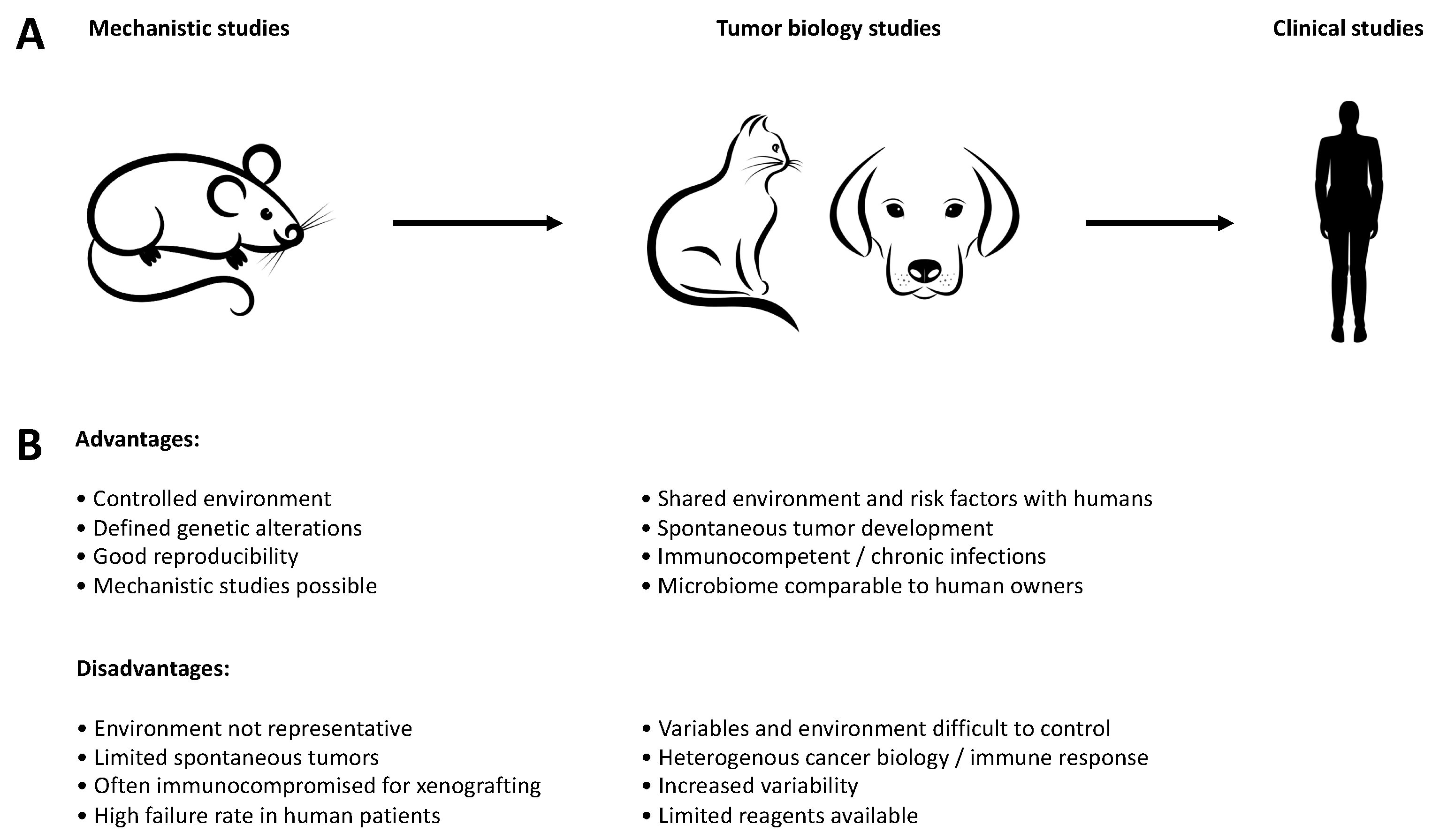
2. Preclinical Models
3. Advantages and Disadvantages of Canine Tumor Models
4. Relevance and Conservation of the JAK-STAT Signaling Pathway
5. Inhibition of STAT3 and STAT5 in Companion Animals: Current Status/Future Perspectives
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Vail, D.M.; MacEwen, E.G. Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Invest. 2000, 18, 781–792. [Google Scholar] [CrossRef]
- Bonnett, B.N.; Egenvall, A.; Hedhammar, A.; Olson, P. Mortality in over 350,000 insured Swedish dogs from 1995-2000: I. Breed-, gender-, age- and cause-specific rates. Acta Vet. Scand. 2005, 46, 105–120. [Google Scholar] [CrossRef]
- Egenvall, A.; Bonnett, B.N.; Hedhammar, A.; Olson, P. Mortality in over 350,000 insured Swedish dogs from 1995-2000: II. Breed-specific age and survival patterns and relative risk for causes of death. Acta Vet. Scand. 2005, 46, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, M.; Baioni, E.; Ru, G.; Carminato, A.; Mutinelli, F. Animal tumour registry of two provinces in northern Italy: Incidence of spontaneous tumours in dogs and cats. BMC Vet. Res. 2009, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Withrow, S.J.; Vail, D.M. Withrow & MacEwen’s Small Animal Clinical Oncology, 5th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2013. [Google Scholar]
- Paoloni, M.; Khanna, C. Translation of new cancer treatments from pet dogs to humans. Nat. Rev. Cancer 2008, 8, 147–156. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Surveillance, Epidemiology and End Results (SEER) Program 2012–2016 Data. Available online: www.seer.cancer.gov/statfacts (accessed on 6 November 2019).
- Vascellari, M.; Capello, K.; Carminato, A.; Zanardello, C.; Baioni, E.; Mutinelli, F. Incidence of mammary tumors in the canine population living in the Veneto region (Northeastern Italy): Risk factors and similarities to human breast cancer. Prev. Vet. Med. 2016, 126, 183–189. [Google Scholar] [CrossRef]
- Hassan, B.B.; Elshafae, S.M.; Supsavhad, W.; Simmons, J.K.; Dirksen, W.P.; Sokkar, S.M.; Rosol, T.J. Feline Mammary Cancer. Vet. Pathol. 2017, 54, 32–43. [Google Scholar] [CrossRef]
- Graf, R.; Pospischil, A.; Guscetti, F.; Meier, D.; Welle, M.; Dettwiler, M. Cutaneous tumors in Swiss dogs: Retrospective data from the Swiss canine cancer registry, 2008–2013. Vet. Pathol. 2018, 55, 809–820. [Google Scholar] [CrossRef]
- Merlo, D.F.; Rossi, L.; Pellegrino, C.; Ceppi, M.; Cardellino, U.; Capurro, C.; Ratto, A.; Sambucco, P.L.; Sestito, V.; Tanara, G.; et al. Cancer incidence in pet dogs: Findings of the animal tumor registry of Genoa, Italy. J. Vet. Intern. Med. 2008, 22, 976–984. [Google Scholar] [CrossRef]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Pinello, K.C.; Niza-Ribeiro, J.; Fonseca, L.; de Matos, A.J. Incidence, characteristics and geographical distributions of canine and human non-Hodgkin’s lymphoma in the Porto region (North West Portugal). Vet. J. 2019, 245, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Grover, S. Gastronintestinal lymphoma in cats. Oncol. Compend. 2005, 27, 741–751. [Google Scholar]
- Egenvall, A.; Nodtvedt, A.; von Euler, H. Bone tumors in a population of 400 000 insured Swedish dogs up to 10 y of age: Incidence and survival. Can. J. Vet. Res. 2007, 71, 292–299. [Google Scholar] [PubMed]
- Dimopoulou, M.; Kirpensteijn, J.; Moens, H.; Kik, M. Histologic prognosticators in feline osteosarcoma: A comparison with phenotypically similar canine osteosarcoma. Vet. Surg. 2008, 37, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Orsulic, S. Mouse models of cancer. Annu. Rev. Pathol 2011, 6, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Ghebranious, N.; Donehower, L.A. Mouse models in tumor suppression. Oncogene 1998, 17, 3385–3400. [Google Scholar] [CrossRef] [PubMed]
- Fowlis, D.J.; Balmain, A. Oncogenes and tumour suppressor genes in transgenic mouse models of neoplasia. Eur. J. Cancer 1993, 29A, 638–645. [Google Scholar] [CrossRef]
- Murphy, W.J. Of mice and men. Biol. Blood Marrow Transplant. 2013, 19, 1140–1141. [Google Scholar] [CrossRef][Green Version]
- Cekanova, M.; Rathore, K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Des. Devel. Ther. 2014, 8, 1911–1921. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 Activation in Solid Cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef] [PubMed]
- Logotheti, S.; Putzer, B.M. STAT3 and STAT5 targeting for simultaneous management of melanoma and autoimmune diseases. Cancers 2019, 11, 1448. [Google Scholar] [CrossRef] [PubMed]
- Rebe, C.; Ghiringhelli, F. STAT3, a master regulator of anti-tumor immune response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Invest. 2002, 109, 1139–1142. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Fagard, R.; Metelev, V.; Souissi, I.; Baran-Marszak, F. STAT3 inhibitors for cancer therapy: Have all roads been explored? JAKSTAT 2013, 2, e22882. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef]
- Dobrowolski, P.; Fischer, M.; Naumann, R. Novel insights into the genetic background of genetically modified mice. Transgenic Res. 2018, 27, 265–275. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Bouchlaka, M.N.; Murphy, W.J. Impact of aging in cancer immunotherapy: The importance of using accurate preclinical models. Oncoimmunology 2013, 2, e27186. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Honda, K. Intestinal commensal microbes as immune modulators. Cell Host Microbe 2012, 12, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Antonovic, L.; Martinez, M. Role of the cytochrome P450 enzyme system in veterinary pharmacokinetics: Where are we now? Where are we going? Future Med. Chem. 2011, 3, 855–879. [Google Scholar] [CrossRef] [PubMed]
- Brekke, T.D.; Steele, K.A.; Mulley, J.F. Inbred or outbred? Genetic diversity in laboratory rodent colonies. G3 (Bethesda) 2018, 8, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Cutler, G.; Kassner, P.D. Copy number variation in the mouse genome: Implications for the mouse as a model organism for human disease. Cytogenet. Genome Res. 2008, 123, 297–306. [Google Scholar] [CrossRef]
- Hartman, J.L.t.; Garvik, B.; Hartwell, L. Principles for the buffering of genetic variation. Science 2001, 291, 1001–1004. [Google Scholar] [CrossRef]
- Srivastava, A.; Morgan, A.P.; Najarian, M.L.; Sarsani, V.K.; Sigmon, J.S.; Shorter, J.R.; Kashfeen, A.; McMullan, R.C.; Williams, L.H.; Giusti-Rodriguez, P.; et al. Genomes of the mouse collaborative cross. Genetics 2017, 206, 537–556. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Casanova, I.; Parreno, M.; Mangues, R. Mouse models in oncogenesis and cancer therapy. Clin. Transl. Oncol. 2006, 8, 318–329. [Google Scholar] [CrossRef]
- Holzapfel, B.M.; Wagner, F.; Thibaudeau, L.; Levesque, J.P.; Hutmacher, D.W. Concise review: Humanized models of tumor immunology in the 21st century: Convergence of cancer Research and tissue engineering. Stem Cells 2015, 33, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.; Su, Y. Mouse xenograft models vs. GEM models for human cancer therapeutics. Dis Model. Mech. 2008, 1, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L. Persistence in the population: Epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein-Barr virus epidemiology, serology, and genetic variability of LMP-1 oncogene among healthy population: An update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Colugnati, F.A.; Staras, S.A.; Dollard, S.C.; Cannon, M.J. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect. Dis 2007, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Willyard, C. The mice with human tumours: Growing pains for a popular cancer model. Nature 2018, 560, 156–157. [Google Scholar] [CrossRef]
- Ireson, C.R.; Alavijeh, M.S.; Palmer, A.M.; Fowler, E.R.; Jones, H.J. The role of mouse tumour models in the discovery and development of anticancer drugs. Br. J. Cancer 2019, 121, 101–108. [Google Scholar] [CrossRef]
- Ciociola, A.A.; Cohen, L.B.; Kulkarni, P.; Gastroenterology, F.D.-R.M.C.o.t.A.C.o. How drugs are developed and approved by the FDA: Current process and future directions. Am. J. Gastroenterol. 2014, 109, 620–623. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Breen, M. Comparative oncology: What dogs and other species can teach us about humans with cancer. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef]
- Hahn, K.A.; Bravo, L.; Adams, W.H.; Frazier, D.L. Naturally occurring tumors in dogs as comparative models for cancer therapy research. In Vivo 1994, 8, 133–143. [Google Scholar] [PubMed]
- Reif, J.S.; Bruns, C.; Lower, K.S. Cancer of the nasal cavity and paranasal sinuses and exposure to environmental tobacco smoke in pet dogs. Am. J. Epidemiol. 1998, 147, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Reif, J.S.; Dunn, K.; Ogilvie, G.K.; Harris, C.K. Passive smoking and canine lung cancer risk. Am. J. Epidemiol. 1992, 135, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, D.; Izquierdo, O.; Eirmann, L.; Binder, S. Myths and misperceptions about ingredients used in commercial pet foods. Vet. Clin. North. Am. Small Anim. Pract. 2014, 44, 689–698. [Google Scholar] [CrossRef]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. Elife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Vonholdt, B.M.; Pollinger, J.P.; Lohmueller, K.E.; Han, E.; Parker, H.G.; Quignon, P.; Degenhardt, J.D.; Boyko, A.R.; Earl, D.A.; Auton, A.; et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 2010, 464, 898–902. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Birney, E. Estimating the neutral rate of nucleotide substitution using introns. Mol. Biol Evol 2007, 24, 522–531. [Google Scholar] [CrossRef]
- Paoloni, M.C.; Khanna, C. Comparative oncology today. Vet. Clin. North. Am. Small Anim Pract 2007, 37, 1023–1032. [Google Scholar] [CrossRef]
- Khanna, C.; Lindblad-Toh, K.; Vail, D.; London, C.; Bergman, P.; Barber, L.; Breen, M.; Kitchell, B.; McNeil, E.; Modiano, J.F.; et al. The dog as a cancer model. Nat. Biotechnol. 2006, 24, 1065–1066. [Google Scholar] [CrossRef]
- Di Cerbo, A.; Palmieri, B.; De Vico, G.; Iannitti, T. Onco-epidemiology of domestic animals and targeted therapeutic attempts: Perspectives on human oncology. J. Cancer Res. Clin. Oncol 2014, 140, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Modiano, J.F.; Breen, M.; Burnett, R.C.; Parker, H.G.; Inusah, S.; Thomas, R.; Avery, P.R.; Lindblad-Toh, K.; Ostrander, E.A.; Cutter, G.C.; et al. Distinct B-cell and T-cell lymphoproliferative disease prevalence among dog breeds indicates heritable risk. Cancer Res. 2005, 65, 5654–5661. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, A.T.; Massoco, C.O.; Felizzola, C.R.; Perlmann, E.; Batschinski, K.; Tedardi, M.V.; Garcia, J.S.; Mendonca, P.P.; Teixeira, T.F.; Zaidan Dagli, M.L. Comparative aspects of canine melanoma. Vet. Sci. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.M. Breed-predispositions to cancer in pedigree dogs. ISRN Vet. Sci 2013, 2013, 941275. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, M.; Howard, J.; Rossetti, M.; Geissbuhler, U. Life expectancy and causes of death in Bernese mountain dogs in Switzerland. BMC Vet. Res. 2016, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.W.; Wiles, B.M.; Llewellyn-Zaidi, A.M.; Evans, K.M.; O’Neill, D.G. Longevity and mortality in Kennel Club registered dog breeds in the UK in 2014. Canine Genet. Epidemiol. 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.; Hoather, T.; McKinley, T.J.; Wood, J.L. Mortality in a cohort of flat-coated retrievers in the UK. Vet. Comp. Oncol. 2009, 7, 115–121. [Google Scholar] [CrossRef]
- Zink, M.C.; Farhoody, P.; Elser, S.E.; Ruffini, L.D.; Gibbons, T.A.; Rieger, R.H. Evaluation of the risk and age of onset of cancer and behavioral disorders in gonadectomized Vizslas. J. Am. Vet. Med. Assoc. 2014, 244, 309–319. [Google Scholar] [CrossRef]
- Henry, C.J.; Higginbotham, H.M. Cancer Management in Small Animal Practice; Saunders Elsevier: Philadelphia, PA, USA, 2009. [Google Scholar]
- Fan, T.M.; Khanna, C. Comparative Aspects of Osteosarcoma Pathogenesis in Humans and Dogs. Vet. Sci 2015, 2, 210–230. [Google Scholar] [CrossRef]
- Urfer, S.R.; Gaillard, C.; Steiger, A. Lifespan and disease predispositions in the Irish Wolfhound: A review. Vet. Q. 2007, 29, 102–111. [Google Scholar] [CrossRef]
- Zandvliet, M. Canine lymphoma: A review. Vet. Q. 2016, 36, 76–104. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Adaptive immune resistance: How cancer protects from immune attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Goulart, M.R.; Pluhar, G.E.; Ohlfest, J.R. Identification of myeloid derived suppressor cells in dogs with naturally occurring cancer. PLoS ONE 2012, 7, e33274. [Google Scholar] [CrossRef]
- Pinheiro, D.; Chang, Y.M.; Bryant, H.; Szladovits, B.; Dalessandri, T.; Davison, L.J.; Yallop, E.; Mills, E.; Leo, C.; Lara, A.; et al. Dissecting the regulatory microenvironment of a large animal model of non-Hodgkin lymphoma: Evidence of a negative prognostic impact of FOXP3+ T cells in canine B cell lymphoma. PLoS ONE 2014, 9, e105027. [Google Scholar] [CrossRef]
- Coy, J.; Caldwell, A.; Chow, L.; Guth, A.; Dow, S. PD-1 expression by canine T cells and functional effects of PD-1 blockade. Vet. Comp. Oncol. 2017, 15, 1487–1502. [Google Scholar] [CrossRef]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef]
- Reardon, S. CRISPR gene-editing creates wave of exotic model organisms. Nature 2019, 568, 441–442. [Google Scholar] [CrossRef]
- Simoff, I.; Karlgren, M.; Backlund, M.; Lindstrom, A.C.; Gaugaz, F.Z.; Matsson, P.; Artursson, P. Complete knockout of endogenous Mdr1 (Abcb1) in MDCK cells by CRISPR-Cas9. J. Pharm. Sci. 2016, 105, 1017–1021. [Google Scholar] [CrossRef]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef]
- Liongue, C.; Ward, A.C. Evolution of the JAK-STAT pathway. JAKSTAT 2013, 2, e22756. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Polak, K.L.; Chernosky, N.M.; Smigiel, J.M.; Tamagno, I.; Jackson, M.W. Balancing STAT Activity as a Therapeutic Strategy. Cancers 2019, 11, 1716. [Google Scholar] [CrossRef] [PubMed]
- Yue-Ting, K.L.; Ramaiyer, M.; E. Johnson, D.; R. Grandis, J. Targeting STAT3 in cancer with nucleotide therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Meier, J.A.; Larner, A.C. Toward a new STATe: The role of STATs in mitochondrial function. Semin. Immunol. 2014, 26, 20–28. [Google Scholar] [CrossRef]
- Frank, D.A. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007, 251, 199–210. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Nelson, E.A.; Xiang, M.; Frank, D.A. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT 2012, 1, 55–64. [Google Scholar] [CrossRef]
- Roeser, J.C.; Leach, S.D.; McAllister, F. Emerging strategies for cancer immunoprevention. Oncogene 2015, 34, 6029–6039. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kwok, S.K.; Lim, M.A.; Kim, E.K.; Ryu, J.G.; Kim, S.M.; Oh, H.J.; Ju, J.H.; Park, S.H.; Kim, H.Y.; et al. STA-21, a promising STAT-3 inhibitor that reciprocally regulates Th17 and Treg cells, inhibits osteoclastogenesis in mice and humans and alleviates autoimmune inflammation in an experimental model of rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Kunigal, S.; Lakka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar] [PubMed]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; Moriggl, R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim. Biophys. Acta 2011, 1815, 104–114. [Google Scholar] [CrossRef]
- Zhang, H.F.; Lai, R. STAT3 in Cancer-Friend or Foe? Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef]
- Starr, R.; Hilton, D.J. Negative regulation of the JAK/STAT pathway. Bioessays 1999, 21, 47–52. [Google Scholar] [CrossRef]
- Wake, M.S.; Watson, C.J. STAT3 the oncogene - still eluding therapy? FEBS J. 2015, 282, 2600–2611. [Google Scholar] [CrossRef]
- Carmi-Levy, I.; Homey, B.; Soumelis, V. A modular view of cytokine networks in atopic dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 245–253. [Google Scholar] [CrossRef]
- Brandt, E.B.; Sivaprasad, U. Th2 Cytokines and Atopic Dermatitis. J. Clin. Cell Immunol 2011, 2. [Google Scholar] [CrossRef]
- Ong, P.Y.; Leung, D.Y. Immune dysregulation in atopic dermatitis. Curr. Allergy Asthma Rep. 2006, 6, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.B.; Lo, A.; Eden, C.A.; Huntley, S.; Morey, V.; Ramsey, S.; Richardson, C.; Smith, D.J.; Sutton, C.; Taylor, M.D.; et al. Survey of the prevalence, diagnosis and treatment of dermatological conditions in small animals in general practice. Vet. Rec. 2006, 158, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Olivry, T.; Bizikova, P. A systematic review of randomized controlled trials for prevention or treatment of atopic dermatitis in dogs: 2008-2011 update. Vet. Dermatol. 2013, 24, 97–117, e25–e26. [Google Scholar] [CrossRef] [PubMed]
- Marsella, R.; Olivry, T.; Maeda, S. Cellular and cytokine kinetics after epicutaneous allergen challenge (atopy patch testing) with house dust mites in high-IgE beagles. Vet. Dermatol. 2006, 17, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, T.J.; Knight, P.A.; McAleese, S.M.; Lamb, J.R.; Hill, P.B. Expression of Th1, Th2 and immunosuppressive cytokine gene transcripts in canine atopic dermatitis. Clin. Exp. Allergy 2002, 32, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Schlotter, Y.M.; Rutten, V.P.; Riemers, F.M.; Knol, E.F.; Willemse, T. Lesional skin in atopic dogs shows a mixed Type-1 and Type-2 immune responsiveness. Vet. Immunol. Immunopathol. 2011, 143, 20–26. [Google Scholar] [CrossRef]
- Gonzales, A.J.; Bowman, J.W.; Fici, G.J.; Zhang, M.; Mann, D.W.; Mitton-Fry, M. Oclacitinib (APOQUEL((R))) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J. Vet. Pharmacol. Ther. 2014, 37, 317–324. [Google Scholar] [CrossRef]
- Cosgrove, S.B.; Wren, J.A.; Cleaver, D.M.; Walsh, K.F.; Follis, S.I.; King, V.I.; Tena, J.K.; Stegemann, M.R. A blinded, randomized, placebo-controlled trial of the efficacy and safety of the Janus kinase inhibitor oclacitinib (Apoquel(R)) in client-owned dogs with atopic dermatitis. Vet. Dermatol. 2013, 24, 587–597, e141–e142. [Google Scholar] [CrossRef]
- Cosgrove, S.B.; Wren, J.A.; Cleaver, D.M.; Martin, D.D.; Walsh, K.F.; Harfst, J.A.; Follis, S.L.; King, V.L.; Boucher, J.F.; Stegemann, M.R. Efficacy and safety of oclacitinib for the control of pruritus and associated skin lesions in dogs with canine allergic dermatitis. Vet. Dermatol. 2013, 24, 479-e114. [Google Scholar] [CrossRef]
- Collard, W.T.; Hummel, B.D.; Fielder, A.F.; King, V.L.; Boucher, J.F.; Mullins, M.A.; Malpas, P.B.; Stegemann, M.R. The pharmacokinetics of oclacitinib maleate, a Janus kinase inhibitor, in the dog. J. Vet. Pharmacol Ther 2014, 37, 279–285. [Google Scholar] [CrossRef]
- Lu, Z.; Hong, C.C.; Jark, P.C.; Assumpcao, A.; Bollig, N.; Kong, G.; Pan, X. JAK1/2 Inhibitors AZD1480 and CYT387 inhibit canine B-Cell lymphoma growth by increasing apoptosis and disrupting cell proliferation. J. Vet. Intern. Med. 2017, 31, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Assumpcao, A.; Jark, P.C.; Hong, C.C.; Lu, Z.; Ruetten, H.M.; Heaton, C.M.; Pinkerton, M.E.; Pan, X. STAT3 Expression and Activity are Up-Regulated in Diffuse Large B Cell Lymphoma of Dogs. J. Vet. Intern. Med. 2018, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Krol, M.; Pawlowski, K.M.; Dolka, I.; Musielak, O.; Majchrzak, K.; Mucha, J.; Motyl, T. Density of Gr1-positive myeloid precursor cells, p-STAT3 expression and gene expression pattern in canine mammary cancer metastasis. Vet. Res. Commun. 2011, 35, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Van Garderen, E.; Swennenhuis, J.F.; Hellmen, E.; Schalken, J.A. Growth hormone induces tyrosyl phosphorylation of the transcription factors Stat5a and Stat5b in CMT-U335 canine mammary tumor cells. Domest. Anim. Endocrinol. 2001, 20, 123–135. [Google Scholar] [CrossRef]
- Brown, M.E.; Bear, M.D.; Rosol, T.J.; Premanandan, C.; Kisseberth, W.C.; London, C.A. Characterization of STAT3 expression, signaling and inhibition in feline oral squamous cell carcinoma. BMC Vet. Res. 2015, 11, 206. [Google Scholar] [CrossRef]
- Petterino, C.; Podesta, G.; Ratto, A.; Drigo, M.; Pellegrino, C. Immunohistochemical study of phospho-Stat3-ser727 expression in feline mammary gland tumours. Vet. Res. Commun. 2007, 31, 173–184. [Google Scholar] [CrossRef]
- Petterino, C.; Ratto, A.; Podesta, G.; Drigo, M.; Pellegrino, C. Immunohistochemical evaluation of STAT3-p-tyr705 expression in feline mammary gland tumours and correlation with histologic grade. Res. Vet. Sci. 2007, 82, 218–224. [Google Scholar] [CrossRef]
- Willmann, M.; Hadzijusufovic, E.; Hermine, O.; Dacasto, M.; Marconato, L.; Bauer, K.; Peter, B.; Gamperl, S.; Eisenwort, G.; Jensen-Jarolim, E.; et al. Comparative oncology: The paradigmatic example of canine and human mast cell neoplasms. Vet. Comp. Oncol. 2019, 17, 1–10. [Google Scholar] [CrossRef]
- Makielski, K.M.; Mills, L.J.; Sarver, A.L.; Henson, M.S.; Spector, L.G.; Naik, S.; Modiano, J.F. Risk factors for development of canine and human osteosarcoma: A comparative review. Vet. Sci. 2019, 6, 48. [Google Scholar] [CrossRef]
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the surveillance, epidemiology, and end results program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Fenger, J.M.; London, C.A.; Kisseberth, W.C. Canine osteosarcoma: A naturally occurring disease to inform pediatric oncology. ILAR J. 2014, 55, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.L.; Liao, A.T.; McCleese, J.K.; Bear, M.D.; Lin, J.; Li, P.K.; Kisseberth, W.C.; London, C.A. Characterization of STAT3 activation and expression in canine and human osteosarcoma. BMC Cancer 2009, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 2011, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Couto, J.I.; Bear, M.D.; Lin, J.; Pennel, M.; Kulp, S.K.; Kisseberth, W.C.; London, C.A. Biologic activity of the novel small molecule STAT3 inhibitor LLL12 against canine osteosarcoma cell lines. BMC Vet. Res. 2012, 8, 244. [Google Scholar] [CrossRef] [PubMed]
- Onimoe, G.I.; Liu, A.; Lin, L.; Wei, C.C.; Schwartz, E.B.; Bhasin, D.; Li, C.; Fuchs, J.R.; Li, P.K.; Houghton, P.; et al. Small molecules, LLL12 and FLLL32, inhibit STAT3 and exhibit potent growth suppressive activity in osteosarcoma cells and tumor growth in mice. Invest. New Drugs 2012, 30, 916–926. [Google Scholar] [CrossRef]
- Paoloni, M.; Davis, S.; Lana, S.; Withrow, S.; Sangiorgi, L.; Picci, P.; Hewitt, S.; Triche, T.; Meltzer, P.; Khanna, C. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics 2009, 10, 625. [Google Scholar] [CrossRef]
- Rodriguez, C.O., Jr. Using canine osteosarcoma as a model to assess efficacy of novel therapies: Can old dogs teach us new tricks? Adv. Exp. Med. Biol. 2014, 804, 237–256. [Google Scholar] [CrossRef]
- London, C.A.; Seguin, B. Mast cell tumors in the dog. Vet. Clin. North. Am. Small Anim. Pract. 2003, 33, 473–489. [Google Scholar] [CrossRef]
- Misdorp, W. Mast cells and canine mast cell tumours. A review. Vet. Q. 2004, 26, 156–169. [Google Scholar] [CrossRef]
- Shoop, S.J.; Marlow, S.; Church, D.B.; English, K.; McGreevy, P.D.; Stell, A.J.; Thomson, P.C.; O’Neill, D.G.; Brodbelt, D.C. Prevalence and risk factors for mast cell tumours in dogs in England. Canine Genet. Epidemiol. 2015, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [PubMed]
- Letard, S.; Yang, Y.; Hanssens, K.; Palmerini, F.; Leventhal, P.S.; Guery, S.; Moussy, A.; Kinet, J.P.; Hermine, O.; Dubreuil, P. Gain-of-function mutations in the extracellular domain of KIT are common in canine mast cell tumors. Mol. Cancer Res. 2008, 6, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- London, C.A.; Galli, S.J.; Yuuki, T.; Hu, Z.Q.; Helfand, S.C.; Geissler, E.N. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp. Hematol. 1999, 27, 689–697. [Google Scholar] [CrossRef]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar] [CrossRef]
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef]
- Iemura, A.; Tsai, M.; Ando, A.; Wershil, B.K.; Galli, S.J. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J. Pathol. 1994, 144, 321–328. [Google Scholar]
- Moller, C.; Alfredsson, J.; Engstrom, M.; Wootz, H.; Xiang, Z.; Lennartsson, J.; Jonsson, J.I.; Nilsson, G. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood 2005, 106, 1330–1336. [Google Scholar] [CrossRef]
- Hahn, K.A.; Ogilvie, G.; Rusk, T.; Devauchelle, P.; Leblanc, A.; Legendre, A.; Powers, B.; Leventhal, P.S.; Kinet, J.P.; Palmerini, F.; et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J. Vet. Intern. Med. 2008, 22, 1301–1309. [Google Scholar] [CrossRef]
- London, C.A.; Malpas, P.B.; Wood-Follis, S.L.; Boucher, J.F.; Rusk, A.W.; Rosenberg, M.P.; Henry, C.J.; Mitchener, K.L.; Klein, M.K.; Hintermeister, J.G.; et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin. Cancer Res. 2009, 15, 3856–3865. [Google Scholar] [CrossRef]
- Chaix, A.; Lopez, S.; Voisset, E.; Gros, L.; Dubreuil, P.; De Sepulveda, P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J. Biol. Chem. 2011, 286, 5956–5966. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, C.; Cerny-Reiterer, S.; Sonneck, K.; Mayerhofer, M.; Gleixner, K.V.; Fritz, R.; Kerenyi, M.; Boudot, C.; Gouilleux, F.; Kornfeld, J.W.; et al. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: Subcellular distribution and role of the transforming oncoprotein KIT D816V. Am. J. Pathol. 2009, 175, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Wingelhofer, B.; Peter, B.; Bauer, K.; Berger, D.; Gamperl, S.; Reifinger, M.; Cerny-Reiterer, S.; Moriggl, R.; Willmann, M.; et al. The JAK2/STAT5 signaling pathway as a potential therapeutic target in canine mastocytoma. Vet. Comp. Oncol. 2018, 16, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.; Bibi, S.; Eisenwort, G.; Wingelhofer, B.; Berger, D.; Stefanzl, G.; Blatt, K.; Herrmann, H.; Hadzijusufovic, E.; Hoermann, G.; et al. Drug-induced inhibition of phosphorylation of STAT5 overrides drug resistance in neoplastic mast cells. Leukemia 2018, 32, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Page, B.D.; Khoury, H.; Laister, R.C.; Fletcher, S.; Vellozo, M.; Manzoli, A.; Yue, P.; Turkson, J.; Minden, M.D.; Gunning, P.T. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J. Med. Chem. 2012, 55, 1047–1055. [Google Scholar] [CrossRef]
- Park, J.S.; Withers, S.S.; Modiano, J.F.; Kent, M.S.; Chen, M.; Luna, J.I.; Culp, W.T.N.; Sparger, E.E.; Rebhun, R.B.; Monjazeb, A.M.; et al. Canine cancer immunotherapy studies: Linking mouse and human. J. Immunother. Cancer 2016, 4, 97. [Google Scholar] [CrossRef]
- Alvarez, C.E. Naturally occurring cancers in dogs: Insights for translational genetics and medicine. ILAR J. 2014, 55, 16–45. [Google Scholar] [CrossRef]
- Furdos, I.; Fazekas, J.; Singer, J.; Jensen-Jarolim, E. Translating clinical trials from human to veterinary oncology and back. J. Transl. Med. 2015, 13, 265. [Google Scholar] [CrossRef]
- Riccardo, F.; Aurisicchio, L.; Impellizeri, J.A.; Cavallo, F. The importance of comparative oncology in translational medicine. Cancer Immunol. Immunother. 2015, 64, 137–148. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tissue | Human | Dog | Cat |
|---|---|---|---|
| Mammary | 127.5 [7] | 250 [8] | 13–25 [9] |
| Melanoma | 22.2 [7] | 19.8 [10] | ND |
| Testes | 5.9 [7] | 16.7 [11] | ND |
| Connective Tissue | 3.5 [7] | 40.1 [10] | 17.0 [1] |
| Skin | 98.85 [12] | 103.3 [10] | 34.7 [1] |
| Oral | 11.3 [7] | 20.4 [1] | 11.6 [1] |
| NHL/Leukemia | 33.7 [7] | 76.3 [13] | 41 [14] |
| Bone | 1.0 [7] | 27.2 [15] | 3.1–4.9 [16] |
| Breeds | Most Frequent Tumor Types |
|---|---|
| Bernese Mountain Dog | Histiocytic sarcoma [68], Lymphoma [68,69], Osteosarcoma [68] |
| Boxer | Glioma [67,69], Mast cell tumor [10,67] |
| Flat-Coated Retriever | Soft tissue sarcoma, Histiocytic sarcoma, Hemangiosarcoma [70] |
| Golden Retriever | Mast cell tumor, Lymphoma, Oral Melanoma, Fibrosarcoma [67] |
| Magyar Viszla | Mast cell tumor, Hemangiosarcoma, Lymphoma [10,71] |
| Giant Schnauzer | Epidermal tumor, Hair follicle tumor, Melanocytic tumor [10] |
| Airedale Terrier | Melanoma [72], Lymphoma [67], Prostatic carcinoma [67] |
| Bullmastiff | Mast cell tumor, Lymphoma [67] |
| St. Bernard | Lymphoma [67], Osteosarcoma [73] |
| Irish Wolfhound | Osteosarcoma [67,74], Lymphoma [75] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kieslinger, M.; Swoboda, A.; Kramer, N.; Pratscher, B.; Wolfesberger, B.; Burgener, I.A. Companion Animals as Models for Inhibition of STAT3 and STAT5. Cancers 2019, 11, 2035. https://doi.org/10.3390/cancers11122035
Kieslinger M, Swoboda A, Kramer N, Pratscher B, Wolfesberger B, Burgener IA. Companion Animals as Models for Inhibition of STAT3 and STAT5. Cancers. 2019; 11(12):2035. https://doi.org/10.3390/cancers11122035
Chicago/Turabian StyleKieslinger, Matthias, Alexander Swoboda, Nina Kramer, Barbara Pratscher, Birgitt Wolfesberger, and Iwan A. Burgener. 2019. "Companion Animals as Models for Inhibition of STAT3 and STAT5" Cancers 11, no. 12: 2035. https://doi.org/10.3390/cancers11122035
APA StyleKieslinger, M., Swoboda, A., Kramer, N., Pratscher, B., Wolfesberger, B., & Burgener, I. A. (2019). Companion Animals as Models for Inhibition of STAT3 and STAT5. Cancers, 11(12), 2035. https://doi.org/10.3390/cancers11122035