Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma

Abstract
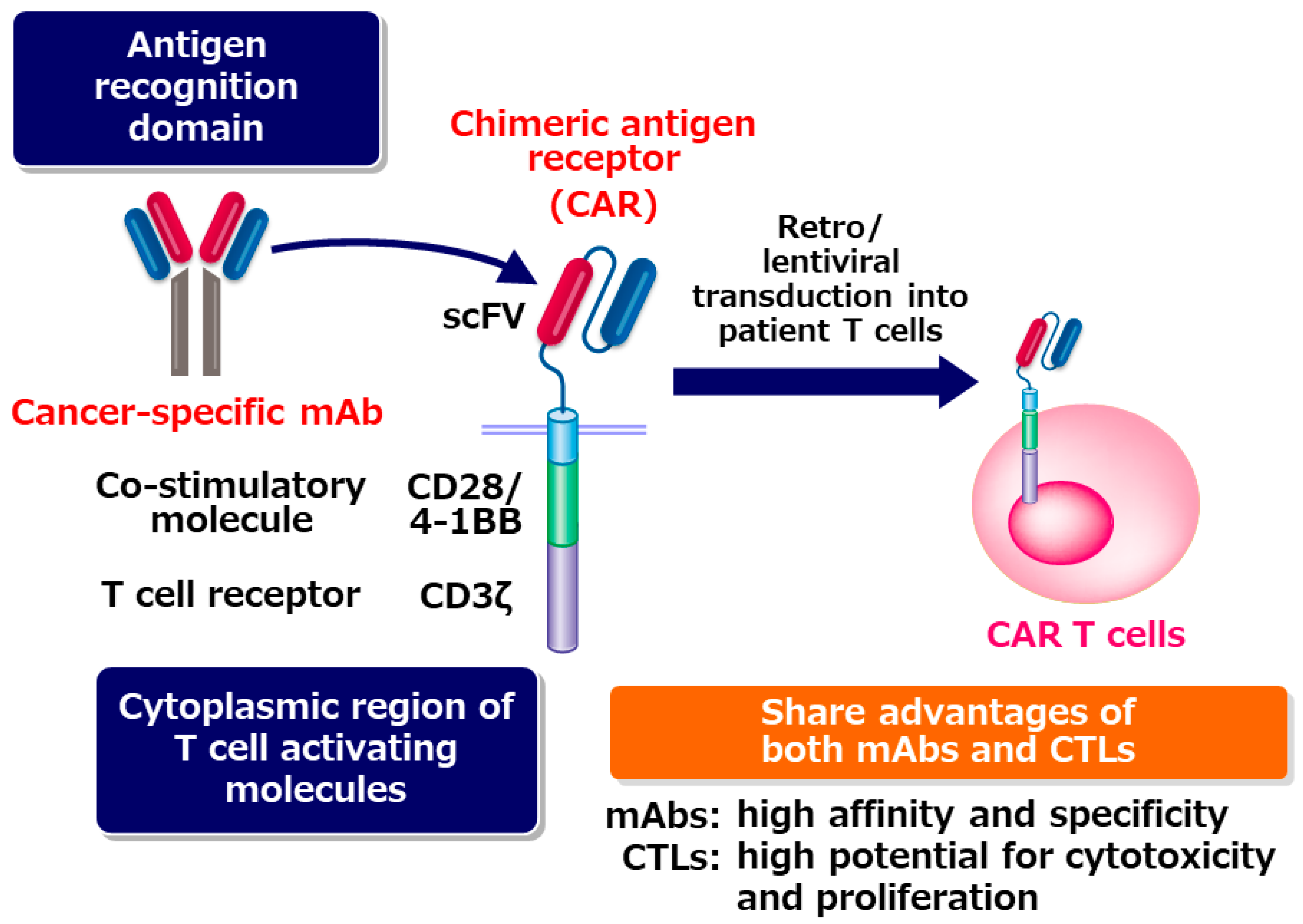
1. Chimeric antigen Receptor (CAR) T-Cell Therapy
2. BCMA CAR T-Cell Therapy for Multiple Myeloma
3. CD19-Positive B Cells as Candidates for Therapeutic Targets to Cure MM
4. Clinical Trial of CAR T-Cell Therapy Targeting CD19
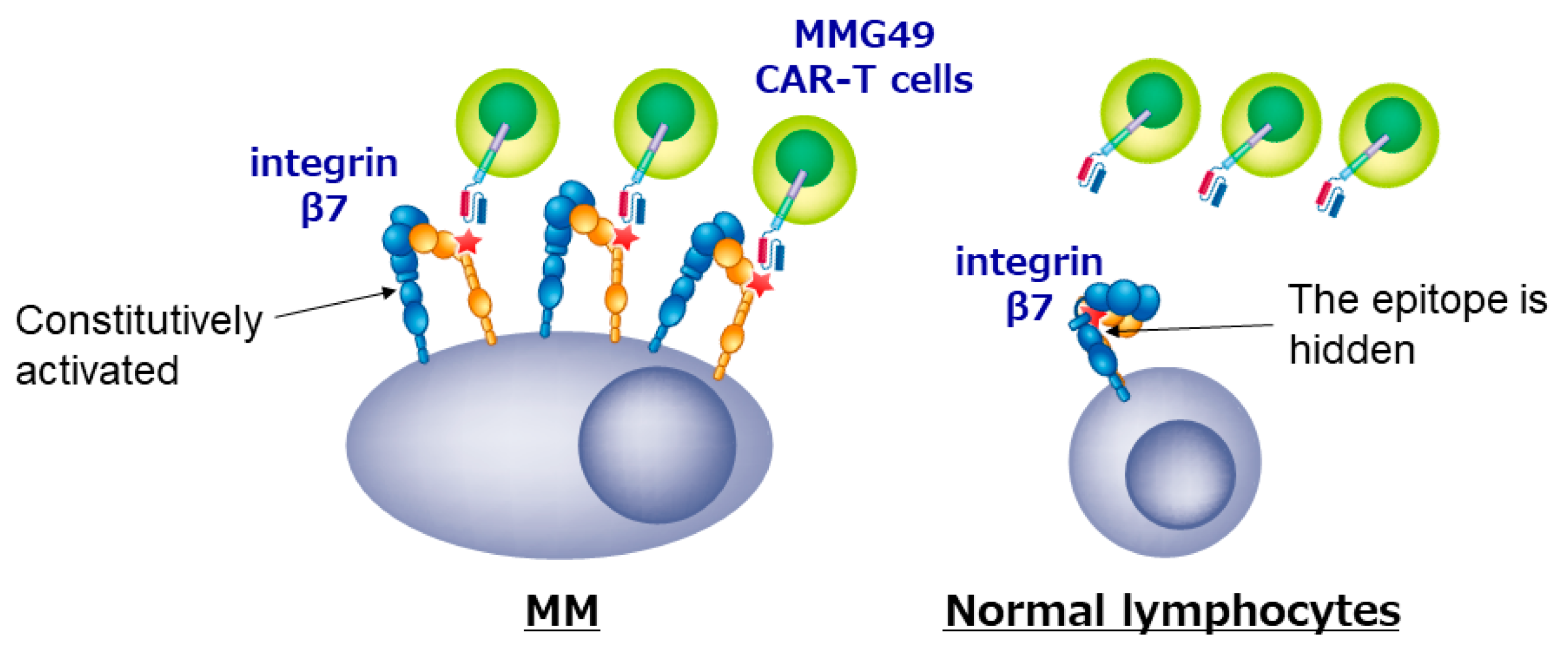
5. Development of CAR T-Cells Targeting Antigens other than BCMA and CD19
6. Future Perspective
6.1. Improvement of Trafficking of CAR T Cells to Tumor Sites
6.2. Enhancement of Persistence of CAR T Cells In Vivo
6.3. Development of CAR T Cell Therapy without CRS
6.4. Development of off the Shelf CAR T Cells
6.5. Armored CAR T Cells
6.6. Avoidance of T Cell Exhaustion
6.7. T Lymphocyte Metabolism
Funding
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.; Yeh, R.; Bernal, Y.; Riviere, I.; Sadelain, M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: Case report of an unforeseen adverse event in a phase I clinical trial. Mol. Ther. 2010, 18, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 130. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, L.J.; Yang, S.S.; Sun, Y.; Wu, W.; Liu, Y.F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.Q.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [PubMed]
- Mailankody, S.; Ghosh, A.; Staehr, M.; Purdon, T.J.; Roshal, M.; Halton, E.; Wills, A. Clinical responses and pharmacokinetics of MCARH171, a human-derived Bcma targeted CAR T cell therapy in relapsed/refractory multiple myeloma: Final results of a phase I clinical trial. Blood 2018, 132 (Suppl. 1), 959. [Google Scholar] [CrossRef]
- Li, C.; Wang, Q.; Zhu, H.; Mao, X.; Wang, Y.; Zhang, Y.; Zhou, J. T cells expressing anti B-cell maturation antigen chimeric antigen receptors for plasma cell malignancies. Blood 2018, 132 (Suppl. 1), 1013. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lopez-Moyado, I.F.; Seo, H.; Lio, C.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.C.; Dundar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Bakkus, M.H.; Heirman, C.; Van Riet, I.; Van Camp, B.; Thielemans, K. Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood 1992, 80, 2326–2335. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Smith, A.M.; Szczepek, A.; Mant, M.J.; Belch, A.R.; Pilarski, L.M. In multiple myeloma, clonotypic B lymphocytes are detectable among CD19+ peripheral blood cells expressing CD38, CD56, and monotypic Ig light chain. Blood 1995, 85, 436–447. [Google Scholar] [CrossRef]
- Billadeau, D.; Ahmann, G.; Greipp, P.; Van Ness, B. The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J. Exp. Med. 1993, 178, 1023–1031. [Google Scholar] [CrossRef]
- Matsui, W.; Wang, Q.; Barber, J.P.; Brennan, S.; Smith, B.D.; Borrello, I.; McNiece, I.; Lin, L.; Ambinder, R.F.; Peacock, C.; et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008, 68, 190–197. [Google Scholar] [CrossRef]
- Hosen, N.; Matsuoka, Y.; Kishida, S.; Nakata, J.; Mizutani, Y.; Hasegawa, K.; Mugitani, A.; Ichihara, H.; Aoyama, Y.; Nishida, S.; et al. CD138-negative clonogenic cells are plasma cells but not B cells in some multiple myeloma patients. Leukemia 2012, 26, 2135–2141. [Google Scholar] [CrossRef]
- Kim, D.; Park, C.Y.; Medeiros, B.C.; Weissman, I.L. CD19-CD45 low/-CD38 high/CD138+plasma cells enrich for human tumorigenic myeloma cells. Leukemia 2012, 26, 2530–2537. [Google Scholar] [CrossRef]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.L.; Stadtmauer, E.A.; June, C.H. Chimeric Antigen Receptor T Cells in Myeloma. N. Engl. J. Med. 2016, 374, 194. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7(+) normal lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef]
- Schmidts, A.; Ormhoj, M.; Choi, B.D.; Taylor, A.O.; Bouffard, A.A.; Scarfo, I.; Larson, R.C.; Frigault, M.J.; Gallagher, K.; Castano, A.P.; et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. 2019, 3, 3248–3260. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S.; et al. The activated conformation of integrin beta7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat. Med. 2017, 23, 1436–1443. [Google Scholar] [CrossRef]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell 2018, 23, 850–858. [Google Scholar] [CrossRef]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8alphabeta T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Muranski, P.; Kaiser, A.; Boni, A.; Sanchez-Perez, L.; Yu, Z.; Palmer, D.C.; Reger, R.N.; Borman, Z.A.; Zhang, L.; et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010, 70, 6725–6734. [Google Scholar] [CrossRef]
- Boice, M.; Salloum, D.; Mourcin, F.; Sanghvi, V.; Amin, R.; Oricchio, E.; Jiang, M.; Mottok, A.; Denis-Lagache, N.; Ciriello, G.; et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell 2016, 167, 405–418. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488. [Google Scholar] [CrossRef]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Trial | Construct | ORR (Optimal Doses) | VGPR/CR (Optimal Doses) | References |
|---|---|---|---|---|
| NCI | Murine, CD28 | 81% (13/16) | 63% (10/16) | [12] |
| UPENN | Human, 4-1BB | 64% (7/11) | 36% (4/11) | [13] |
| Bluebird | Human, 4-1BB | 96% (21/22) | 86% (19/22) | [14] |
| Nanjing Legend Biotech | Murine, 4-1BB | 88.2% (15/17) | 88.2% (15/17) | [15] |
| Memorial Sloan Kettering | Human 4-1BB | 64% | 2/5 ongoing VGPR (7.5, 10 mo) (high does cohort (>450 × 106 cells) | [16] |
| Tongji Hospital of Tongji Medical College | Murine CD28 | 87% | 73% CR | [17] |
| Target | Construct | Clinical trial | Response | References |
|---|---|---|---|---|
| Kappa light chain | CD28 | Done | 5/8: SD | [34] |
| SLAMF7 | CD28 | Not reported yet | - | [35] |
| BCMA/TACI (APRIL) | CD28 | Not reported yet | - | [36] |
| GPRC5D | 4-1BB | Not reported yet | - | [37] |
| Activated integrin β7 | CD28 | Not reported yet | - | [38] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosen, N. Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma. Cancers 2019, 11, 2024. https://doi.org/10.3390/cancers11122024
Hosen N. Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma. Cancers. 2019; 11(12):2024. https://doi.org/10.3390/cancers11122024
Chicago/Turabian StyleHosen, Naoki. 2019. "Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma" Cancers 11, no. 12: 2024. https://doi.org/10.3390/cancers11122024
APA StyleHosen, N. (2019). Chimeric Antigen Receptor T-Cell Therapy for Multiple Myeloma. Cancers, 11(12), 2024. https://doi.org/10.3390/cancers11122024