Novel and Rare Fusion Transcripts Involving Transcription Factors and Tumor Suppressor Genes in Acute Myeloid Leukemia

 ,
,  , , ,
, , , 
 ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. RNA-Seq Cohort Selection
2.2. Identification and Validation of Fusions Genes
2.3. Expression of Genes Involved in Fusions and Frequency of Rearrangments Across Cancers
2.4. Relative Frequency of ZEB2-BCL11B Chimera in Acute Leukemia
2.5. Specific Pattern of Mutations in Patients Carrying the ZEB2-BCL11B Chimera
2.6. BCL11B Protein Expression in AML and Its Transcriptional Signature
2.7. ZEB2-BCL11B Expression Failed to Sustain Self-Renewal of Murine Hematopoietic Stem and Progenitor Cells
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Chromosome Banding Analysis (CBA)
4.3. Fluorescent In Situ Hybridization (FISH)
4.4. Sequencing and Fusion Detection
4.5. RT-PCR, PCR, qPCR and Sanger Sequencing
4.6. Immunohistochemistry
4.7. Gene Expression Profiling (GEP) and SNP-Array
4.8. Retroviral Transduction Assays
4.9. Serial Colony Replating Assay
4.10. Flow Cytometry Analysis
4.11. Immunoblotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Martens, J.H.A.; Stunnenberg, H.G. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett. 2010, 584, 2662–2669. [Google Scholar] [CrossRef]
- Soverini, S.; De Benedittis, C.; Mancini, M.; Martinelli, G. Best Practices in Chronic Myeloid Leukemia Monitoring and Management. Oncologist 2016, 21, 626–633. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Wen, H.; Li, Y.; Malek, S.N.; Kim, Y.C.; Xu, J.; Chen, P.; Xiao, F.; Huang, X.; Zhou, X.; Xuan, Z.; et al. New Fusion Transcripts Identified in Normal Karyotype Acute Myeloid Leukemia. PLoS ONE 2012, 7, e51203. [Google Scholar] [CrossRef]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. NUP98-PHF23 Is a Chromatin-Modifying Oncoprotein That Causes a Wide Array of Leukemias Sensitive to Inhibition of PHD Histone Reader Function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef]
- Togni, M.; Masetti, R.; Pigazzi, M.; Astolfi, A.; Zama, D.; Indio, V.; Serravalle, S.; Manara, E.; Bisio, V.; Rizzari, C.; et al. Identification of the NUP98-PHF23 fusion gene in pediatric cytogenetically normal acute myeloid leukemia by whole-transcriptome sequencing. J. Hematol. Oncol. 2015, 8, 69. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; World Health Organization; International Agency for Research on Cancer. WHO Classification of Tumours of Haematopoietic and lymphoid Tissues; WHO: Geneva, Switzerland, 2018; ISBN 9789283244943. [Google Scholar]
- Riley, D.A.; Tan, F.; Miletich, D.J.; Skidgel, R.A. Chromosomal Localization of the Genes for Human Carboxypeptidase D (CPD) and the Active 50-Kilodalton Subunit of Human Carboxypeptidase N (CPN1). Genomics 1998, 50, 105–108. [Google Scholar] [CrossRef]
- Matsuura, K.; Nakada, C.; Mashio, M.; Narimatsu, T.; Yoshimoto, T.; Tanigawa, M.; Tsukamoto, Y.; Hijiya, N.; Takeuchi, I.; Nomura, T.; et al. Downregulation of SAV1 plays a role in pathogenesis of high-grade clear cell renal cell carcinoma. BMC Cancer 2011, 11, 523. [Google Scholar] [CrossRef] [PubMed]
- Kudo, S.; Fukuda, M. Structural organization of glycophorin A and B genes: Glycophorin B gene evolved by homologous recombination at Alu repeat sequences. Proc. Natl. Acad. Sci. USA 1989, 86, 4619–4623. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y.; Chen, S.-F.; Hsieh, J.-Y.; Chou, F.; Wang, Y.-H.; Lin, W.-T.; Lee, P.-Y.; Yu, Y.-J.; Lin, L.-Y.; Lin, T.-S.; et al. Structural basis of antizyme-mediated regulation of polyamine homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Bonnart, C.; Gérus, M.; Hoareau-Aveilla, C.; Kiss, T.; Caizergues-Ferrer, M.; Henry, Y.; Henras, A.K. Mammalian HCA66 protein is required for both ribosome synthesis and centriole duplication. Nucleic Acids Res. 2012, 40, 6270–6289. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xu, Y.-P.; Li, J.; Duan, S.-S.; Fu, Y.-J.; Zhang, Y.; Zhao, Y.; Qiao, W.-T.; Chen, Q.-M.; Geng, Y.-Q.; et al. Cloning and characterization of a novel intracellular protein p48.2 that negatively regulates cell cycle progression. Int. J. Biochem. Cell Biol. 2009, 41, 2240–2250. [Google Scholar] [CrossRef]
- Hastings, M.L.; Allemand, E.; Duelli, D.M.; Myers, M.P.; Krainer, A.R. Control of Pre-mRNA Splicing by the General Splicing Factors PUF60 and U2AF65. PLoS ONE 2007, 2, e538. [Google Scholar] [CrossRef]
- Waas, W.F.; de Crécy-Lagard, V.; Schimmel, P. Discovery of a gene family critical to wyosine base formation in a subset of phenylalanine-specific transfer RNAs. J. Biol. Chem. 2005, 280, 37616–37622. [Google Scholar] [CrossRef]
- Matsushita, K.; Kitamura, K.; Rahmutulla, B.; Tanaka, N.; Ishige, T.; Satoh, M.; Hoshino, T.; Miyagi, S.; Mori, T.; Itoga, S.; et al. Haploinsufficiency of the c-myc transcriptional repressor FIR as a dominant negative-alternative splicing model, promoted p53-dependent T-cell acute lymphoblastic leukemia progression by activating Notch1. Oncotarget 2015, 6, 5102–5117. [Google Scholar] [CrossRef][Green Version]
- Jayne, S.; Zwartjes, C.G.M.; Van Schaik, F.M.A.; Timmers, H.T.M. Involvement of the SMRT/NCoR–HDAC3 complex in transcriptional repression by the CNOT2 subunit of the human Ccr4–Not complex. Biochem. J. 2006, 398, 461–467. [Google Scholar] [CrossRef]
- Ito, K.; Inoue, T.; Yokoyama, K.; Morita, M.; Suzuki, T.; Yamamoto, T. CNOT2 depletion disrupts and inhibits the CCR4-NOT deadenylase complex and induces apoptotic cell death. Genes to Cells 2011, 16, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Zwartjes, C.G.M.; Jayne, S.; van den Berg, D.L.C.; Timmers, H.T.M. Repression of Promoter Activity by CNOT2, a Subunit of the Transcription Regulatory Ccr4-Not Complex. J. Biol. Chem. 2004, 279, 10848–10854. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mar, B.G.; Zhang, H.; Puram, R.V.; Vazquez, F.; Weir, B.A.; Hahn, W.C.; Ebert, B.; Pellman, D. The EMT regulator ZEB2 is a novel dependency of human and murine acute myeloid leukemia. Blood 2017, 129. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Riedt, T.; Goossens, S.; Carrillo García, C.; Szczepanski, S.; Brandes, M.; Pieters, T.; Dobrosch, L.; Gütgemann, I.; Farla, N.; et al. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood 2017, 129. [Google Scholar] [CrossRef]
- Ha, V.L.; Luong, A.; Li, F.; Casero, D.; Malvar, J.; Kim, Y.M.; Bhatia, R.; Crooks, G.M.; Parekh, C. The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation. Leukemia 2017. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Torkildsen, S.; Gorunova, L.; Beiske, K.; Tjønnfjord, G.E.; Heim, S.; Panagopoulos, I. Novel ZEB2-BCL11B Fusion Gene Identified by RNA-Sequencing in Acute Myeloid Leukemia with t(2;14)(q22;q32). PLoS ONE 2015, 10, e0132736. [Google Scholar] [CrossRef][Green Version]
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Available online: https://cgap.nci.nih.gov/Chromosomes/Mitelman (accessed on 27 January 2019).
- Palka, G.; Calabrese, G.; Fioritoni, G.; Stuppia, L.; Guanciali Franchi, P.; Marino, M.; Antonucci, A.; Spadano, A.; Torlontano, G. Cytogenetic survey of 80 patients with acute nonlymphocytic leukemia. Cancer Genet. Cytogenet. 1992, 59, 45–50. [Google Scholar] [CrossRef]
- Gmidène, A.; Sennana, H.; Wahchi, I.; Youssef, Y.B.; Jeddi, R.; Elloumi, M.; Saad, A. Cytogenetic profile of a large cohort of Tunisian de novo acute myeloid leukemia. Hematology 2012, 17, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Columbano-Green, L.M.; Romain, D.R.; Carter, J.; Crossen, P.E. t(2;14)(q23;q32.3) as the sole abnormality in a patient with acute nonlymphocytic leukemia (FAB-M4). Cancer Genet. Cytogenet. 1990, 48, 255–257. [Google Scholar] [CrossRef]
- Huang, X.; Du, X.; Li, Y. The role of BCL11B in hematological malignancy. Exp. Hematol. Oncol. 2012, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Rubnitz, J.E.; Onciu, M.; Pounds, S.; Shurtleff, S.; Cao, X.; Raimondi, S.C.; Behm, F.G.; Campana, D.; Razzouk, B.I.; Ribeiro, R.C.; et al. Acute mixed lineage leukemia in children: The experience of St Jude Children’s Research Hospital. Blood 2009, 113, 5083–5089. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef]
- Aventín, A.; Sánchez, J.; Nomdedéu, J.F.; Estany, C.; Forcada, P.; La Starza, R.; Mecucci, C. Novel IGHα translocations, t(2;14)(q14.3;q32) and t(14;17)(q32;q21), in B-cell precursor acute lymphoblastic leukemia. Cancer Genet. Cytogenet. 2008, 185, 57–59. [Google Scholar] [CrossRef]
- Inaba, T.; Oku, N.; Gotoh, H.; Murakami, S.; Oku, N.; Itoh, K.; Ura, Y.; Nakanishi, S.; Shimazaki, C.; Nakagawa, M. Philadelphia chromosome positive precursor B-cell acute lymphoblastic leukemia with a translocation t(2;14)(p13;q32). Leukemia 1991, 5, 719–722. [Google Scholar]
- Stengel, A.; Nadarajah, N.; Haferlach, T.; Dicker, F.; Kern, W.; Meggendorfer, M.; Haferlach, C. Detection of recurrent and of novel fusion transcripts in myeloid malignancies by targeted RNA sequencing. Leukemia 2018, 32, 1229–1238. [Google Scholar] [CrossRef]
- Abbas, S.; Sanders, M.A.; Zeilemaker, A.; Geertsma-Kleinekoort, W.M.C.; Koenders, J.E.; Kavelaars, F.G.; Abbas, Z.G.; Mahamoud, S.; Chu, I.W.T.; Hoogenboezem, R.; et al. Integrated genome-wide genotyping and gene expression profiling reveals BCL11B as a putative oncogene in acute myeloid leukemia with 14q32 aberrations. Haematologica 2014, 99, 848–857. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792. [Google Scholar] [CrossRef]
- Iacobucci, I.; Wen, J.; Meggendorfer, M.; Choi, J.K.; Shi, L.; Pounds, S.B.; Carmichael, C.L.; Masih, K.E.; Morris, S.M.; Lindsley, R.C.; et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat. Genet. 2019, 51, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed]
- Mardin, B.R.; Lange, C.; Baxter, J.E.; Hardy, T.; Scholz, S.R.; Fry, A.M.; Schiebel, E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat. Cell Biol. 2010, 12, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Callus, B.A.; Verhagen, A.M.; Vaux, D.L. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J. 2006, 273, 4264–4276. [Google Scholar] [CrossRef]
- Lin, C.; Yang, L.; Rosenfeld, M.G. Molecular Logic Underlying Chromosomal Translocations, Random or Non-Random? Adv. Cancer Res. 2012, 113, 241–279. [Google Scholar]
- Shugay, M.; Ortiz de Mendíbil, I.; Vizmanos, J.L.; Novo, F.J. Genomic Hallmarks of Genes Involved in Chromosomal Translocations in Hematological Cancer. PLoS Comput. Biol. 2012, 8, e1002797. [Google Scholar] [CrossRef]
- Abdelmagid, S.A.; Too, C.K.L. Prolactin and estrogen up-regulate carboxypeptidase-D to promote nitric oxide production and survival of MCF-7 breast cancer cells. Endocrinology 2008, 149, 4821–4828. [Google Scholar] [CrossRef]
- Thomas, L.N.; Merrimen, J.; Bell, D.G.; Rendon, R.; Goffin, V.; Too, C.K.L. Carboxypeptidase-D is elevated in prostate cancer and its anti-apoptotic activity is abolished by combined androgen and prolactin receptor targeting. Prostate 2014, 74, 732–742. [Google Scholar] [CrossRef]
- Jin, T.; Fu, J.; Feng, X.J.; Wang, S.M.; Huang, X.; Zhu, M.H.; Zhang, S.H. SiRNA-targeted carboxypeptidase D inhibits hepatocellular carcinoma growth. Cell Biol. Int. 2013, 37, 929–939. [Google Scholar] [CrossRef]
- Sohn, E.J.; Jung, D.B.; Lee, H.J.; Han, I.; Lee, J.; Lee, H.; Kim, S.H. CNOT2 promotes proliferation and angiogenesis via VEGF signaling in MDA-MB-231 breast cancer cells. Cancer Lett. 2018, 412, 88–98. [Google Scholar] [CrossRef]
- Fenske, T.S.; Pengue, G.; Mathews, V.; Hanson, P.T.; Hamm, S.E.; Riaz, N.; Graubert, T.A. Stem cell expression of the AML1/ETO fusion protein induces a myeloproliferative disorder in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 15184–15189. [Google Scholar] [CrossRef]
- Schessl, C.; Rawat, V.P.S.; Cusan, M.; Deshpande, A.; Kohl, T.M.; Rosten, P.M.; Spiekermann, K.; Humphries, R.K.; Schnittger, S.; Kern, W.; et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J. Clin. Investig. 2005, 115, 2159–2168. [Google Scholar] [CrossRef]
- Simonetti, G.; Padella, A.; do Valle, I.F.; Fontana, M.C.; Fonzi, E.; Bruno, S.; Baldazzi, C.; Guadagnuolo, V.; Manfrini, M.; Ferrari, A.; et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- International Standing Committee on Human Cytogenomic Nomenclature; McGowan-Jordan, J.; Simons, A.; Schmid, M. ISCN: An International System for Human Cytogenomic Nomenclature (2016); Karger: Basel, Switzerland, 2016; ISBN 9783318058574. [Google Scholar]
- Iyer, M.K.; Chinnaiyan, A.M.; Maher, C.A. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics 2011, 27, 2903–2904. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Hormozdiari, F.; Zayed, A.; Giuliany, R.; Ha, G.; Sun, M.G.F.; Griffith, M.; Heravi Moussavi, A.; Senz, J.; Melnyk, N.; et al. deFuse: An Algorithm for Gene Fusion Discovery in Tumor RNA-Seq Data. PLoS Comput. Biol. 2011, 7, e1001138. [Google Scholar] [CrossRef] [PubMed]
- Paciello, G.; Ficarra, E. FuGePrior: A novel gene fusion prioritization algorithm based on accurate fusion structure analysis in cancer RNA-seq samples. BMC Bioinform. 2017, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Zairis, S.; Ficarra, E.; Acquaviva, A.; Wiggins, C.H.; Frattini, V.; Lasorella, A.; Iavarone, A.; Inghirami, G.; Rabadan, R. Pegasus: A comprehensive annotation and prediction tool for detection of driver gene fusions in cancer. BMC Syst. Biol. 2014, 8, 97. [Google Scholar] [CrossRef]
- Shugay, M.; De Mend??bil, I.O.; Vizmanos, J.L.; Novo, F.J. Oncofuse: A computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 2013, 29, 2539–2546. [Google Scholar] [CrossRef]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G.W. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, M.; Viklund, B.; Isaksson, A. Rawcopy: Improved copy number analysis with Affymetrix arrays. Sci. Rep. 2016, 6, 36158. [Google Scholar] [CrossRef] [PubMed]
- Giotopoulos, G.; van der Weyden, L.; Osaki, H.; Rust, A.G.; Gallipoli, P.; Meduri, E.; Horton, S.J.; Chan, W.-I.; Foster, D.; Prinjha, R.K.; et al. A novel mouse model identifies cooperating mutations and therapeutic targets critical for chronic myeloid leukemia progression. J. Exp. Med. 2015, 212, 1551–1569. [Google Scholar] [CrossRef]
- Kern, W.; Voskova, D.; Schoch, C.; Hiddemann, W.; Schnittger, S.; Haferlach, T.; Fonatsch, C.; Haase, D.; Schoch, C.; Hossfeld, D.; et al. Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood 2004, 104, 3078–3085. [Google Scholar] [CrossRef]
- Bene, M.C.; Castoldi, G.; Knapp, W.; Ludwig, W.D.; Matutes, E.; Orfao, A.; van’t Veer, M.B. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 1995, 9, 1783–1786. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| ID | Karyotype Main Clone | Karyotype Second Clone | Karyotype Other Clones | Blasts | WHO Classification | Other Genetic Abnormalities | Phase | Validated fusion(s) |
|---|---|---|---|---|---|---|---|---|
| 59810 | 46,XX,t(2;14)(q21;q32),t(11;12)(p15;q22) [17] | 46,XX [3] | NA | 80% | AML NOS, without maturation | FLT3, TET2 | Diagnosis | 2 |
| 20 | 46,XY,t(6;17)(p21;q11) [20] | NA | NA | 90% | AML NOS, with maturation | NRAS, SRSF2, STAG2, TET2 | Diagnosis | 2 |
| 21 | 46,XY,t(3;12)(p22;q24),+4,-15,+mar [19] | 46,XY [1] | NA | 80% | AML with mutated RUNX1 (provisional entity) | CBL, DNMT3A, IDH2, KDM6A, RUNX1 | Relapse | 1 |
| 32 | 45,XY,der(12)t(12;18)(p13;q12),-18 [12] | 45,XY,t(4;16)(q31;q22),der(12)t(12;18)(p13;q12),-18 [4] | 45,XY,der(6)t(6;12;18)(p21;p13,q12),-18 [3]/46,XY [1] | 80% | AML with MRC | FLT3, WT1 | Relapse | 0 |
| 84 | 47,XX,+8,del(11)(p11p15),t(15;17)(q24q25), inv(16)(p13q22) [20] | NA | NA | 80% | AML with inv(16)(p13.1q22) | CUX1, NOTCH1 | Diagnosis | 1 |
| 68187 | 46,XX,add(8)(p23),der(16)t(1;16)(q11;q11) [18] | 46,XX [2] | NA | 70% | AML with MRC | ETV6, KDM6A | Diagnosis | 1 |
| 63569 | 46,XY [20] | 46,XY,add(10)(p15) [9] | 46,XY,add(10)(p15),t(1;8)(p36;q13) [2] | 70% | AML with mutated NPM1 | DNMT3A, FLT3, IDH2, NPM1 | Relaspe | 0 |
| 125 | 46,XX [11] | 44~47,XX,t(4;17)(p15;q21),del(5)(q13q33),-7,-18,der(X),+1~3mar [9] | NA | 50% | AML with MRC | TP53 | Diagnosis | 1 |
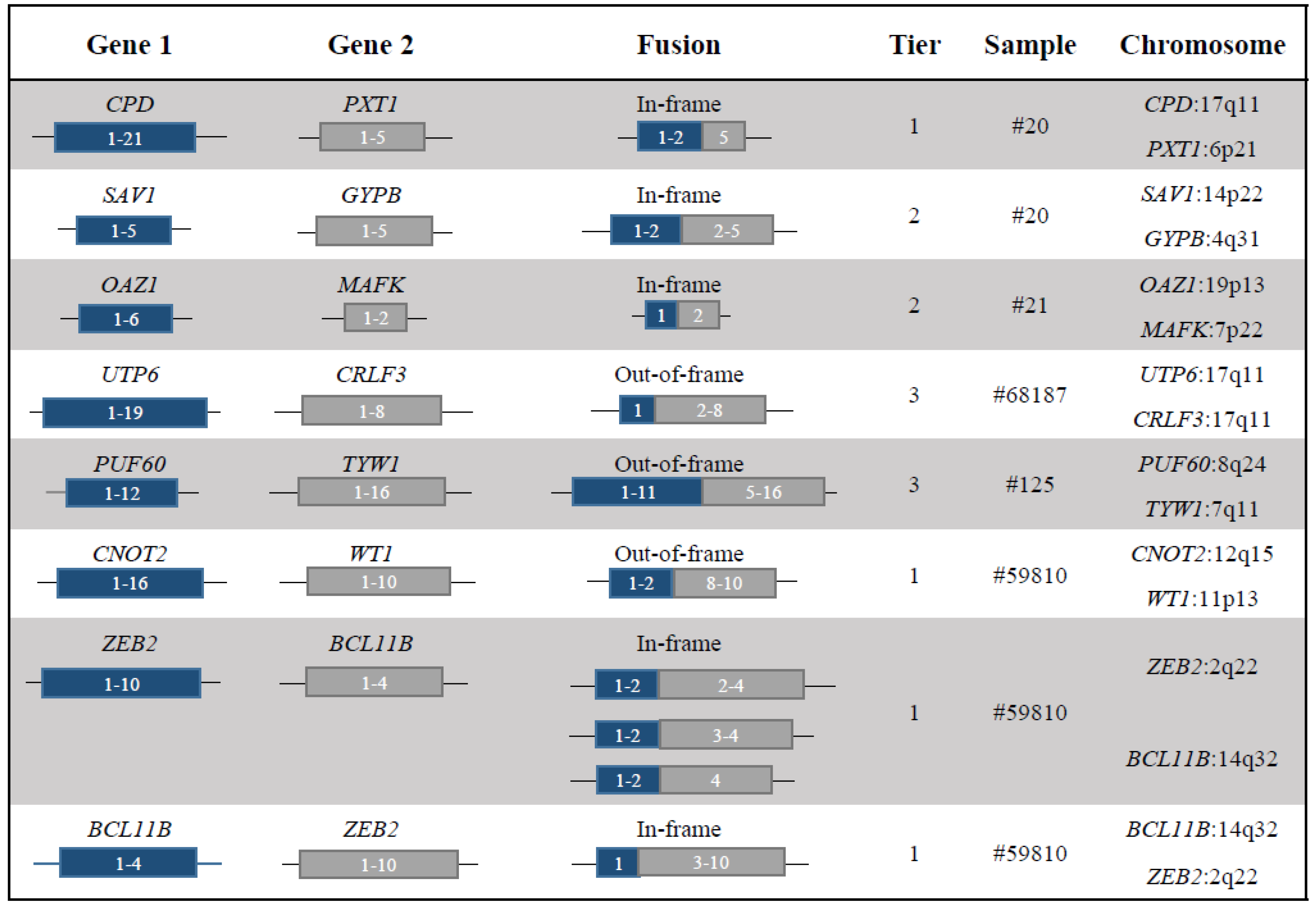
| Sample | Fusion | Gene Function | Category | Fusion Protein Putative Function |
|---|---|---|---|---|
| 20 | CPD-PXT1 | CPD encodes for a metallocarboxypeptidase [11] | NF1 loss | The breakpoint in CPD was associated with a complex rearrangements that involved the loss of NF1. The sample was also characterized by a mutation in NF1 detected by WES. |
| The role of PXT1 is unknown | ||||
| 20 | SAV1-GYPB | SAV1 is a tumor suppressor of the Hippo pathway [12] | Tumor suppressor | Loss of function of SAV1. |
| GYBP is a sialoglycoproteins of the human erythrocyte membrane [13] | ||||
| 21 | OAZ-MAFK | OAZ1 is an Ornithine decarboxylase (ODC) antizyme protein that negatively regulates ODC activity [14] | Transcription factor | The chimera may alter the cellular transcriptional program. |
| MAFK is a transcriptional regulator with bZIP domains [15] | ||||
| 68187 | UTP6-CRLF3 | UTP6 is involved in nucleolar processing of pre-18S ribosomal RNA and centriole duplication [16] | NF1 loss | The rearrangement led to a CN loss involving NF1, which maps in the forward strand of chromosome 17: 29421945-29709134 (GRCh37). |
| CRLF3 is a cytokine receptor-like factor that may negatively regulate cell cycle progression at the G0/G1 phase [17] | ||||
| 125 | PUF60-TYW1 | PUF60 participates in the splicing machinery [18,20] | Tumor suppressor | PUF60 haploinsufficiency was involved in TP53-dependent progression of a T-cell acute lymphoblastic leukaemia [20]. |
| TYW1 may be a component of the wybutosine biosynthesis pathway [19] | ||||
| 59810 | CNOT2-WT1 | CNOT2 encodes for a subunit of the multi-component CCR4-NOT complex, which is involved in transcriptional regulation and mRNA degradation [21,22,23] | Tumor suppressor | The translocation was associated to a deletion at 5’ of WT1, which lead to its CN loss. |
| WT1 is a transcription factor and it is recurrently altered in haematological malignancies, including AML [2] | ||||
| 59810 | ZEB2-BCL11B and BCL11B-ZEB2 | ZEB2 is a transcriptional factor involved in normal and malignant haematopoiesis [24,25] | Transcription factor | The chimera may activate an aberrant transcriptional programme. |
| BCL11B is a transcription factor and key regulator of both differentiation and survival of T-lymphocytes during thymocyte development [26] |
| Case Number | Gender | Age | WHO Classification | Karyotype | FISH | T-cell Markers | BCR | TCR |
|---|---|---|---|---|---|---|---|---|
| 11942 | male | 58 | AML NOS | 46,XY,t(2;14)(q23;q32) | POSITIVE | NA | no clonality detected | no clonality detected |
| 11954 | male | 85 | AML with mutated RUNX1 (provisional entity) | 46,XY,t(2;14)(q14;q32) | POSITIVE | NA | no clonality detected | no clonality detected |
| 11944 | male | 79 | AUL | 46,XY,t(2;14)(q21;q32) | POSITIVE | CD2+; CD7+; TdT+ | clonal | no clonality detected |
| 11945 | male | 59 | T/myeloid MPAL | 46,XY,t(2;14)(q22;q32) | POSITIVE | CD3+; CD7+; CD2+; TdT+ | no clonality detected | clonal |
| 59810 | female | 40 | AML NOS, without maturation | 46,XX,t(2;14)(q21;q32),t(11;12)(p15;q22) | POSITIVE | negative | no clonality detected | no clonality detected |
| A | ||||||||||||
| ASXL1 | BCOR | CALR | CBL | CSF3R | CSNK1A1 | DNMT3A | ETNK1 | ETV6 | EZH2 | FLT3-TKD | ||
| #11942 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | |
| #11944 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | |
| #11954 | NEG | NA | NA | NA | NA | NA | NEG | NA | NA | NA | NEG | |
| #11945 | NEG | NEG | NEG | NEG | NEG | NEG | POS | NEG | NEG | NEG | POS | |
| #59810 | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | POS | |
| B | ||||||||||||
| FLT3-TKD mutation and VAF | FLT3-ITD | FLT3-ITD VAF | GATA1 | GATA2 | IDH1 | IDH2 | JAK2 | |||||
| NEG | NEG | NEG | NEG | NEG | NEG | |||||||
| POS | >0,5 | NEG | NEG | NEG | NEG | NEG | ||||||
| POS | >0,5 | NA | NA | NEG | NEG | POS | ||||||
| c.2516A>G, c.2503G>T; p.Asp839Gly, p.Asp835Tyr; 4%, 8% | POS | <0,5 | NEG | POS | NEG | NEG | NEG | |||||
| c.2516A>G, p.Asp839Gly 34% | POS | <0,5 | NEG | NEG | NEG | NEG | NEG | |||||
| C | ||||||||||||
| KIT | KRAS | MPL | NPM1 | NRAS | PHF6 | PTPN11 | RUNX1 | SETBP1 | SF3B1 | SRSF2 | STAG2 | STAT3 |
| NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG |
| NEG | NEG | NEG | NEG | NEG | NEG | NEG | VARIANTE | VARIANTE | NEG | POS | NA | NEG |
| NA | NEG | NA | NEG | NEG | NA | NA | POS | NA | NEG | NA | NA | NA |
| NEG | NEG | NEG | NEG | NEG | NEG | NEG | VARIANTE | NEG | NEG | NEG | NEG | NA |
| NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG | NEG |
| D | ||||||||||||
| STAT5B | TET2 | TP53 | U2AF1 | WT1 | ZRSR2 | |||||||
| NEG | VARIANTE | NEG | NEG | NEG | NEG | |||||||
| NEG | POS | NEG | NEG | NEG | NEG | |||||||
| NA | NEG | NEG | NA | NA | NA | |||||||
| NA | NEG | NEG | NEG | NEG | NEG | |||||||
| NEG | POS | NEG | NEG | NEG | NEG | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padella, A.; Simonetti, G.; Paciello, G.; Giotopoulos, G.; Baldazzi, C.; Righi, S.; Ghetti, M.; Stengel, A.; Guadagnuolo, V.; De Tommaso, R.; et al. Novel and Rare Fusion Transcripts Involving Transcription Factors and Tumor Suppressor Genes in Acute Myeloid Leukemia. Cancers 2019, 11, 1951. https://doi.org/10.3390/cancers11121951
Padella A, Simonetti G, Paciello G, Giotopoulos G, Baldazzi C, Righi S, Ghetti M, Stengel A, Guadagnuolo V, De Tommaso R, et al. Novel and Rare Fusion Transcripts Involving Transcription Factors and Tumor Suppressor Genes in Acute Myeloid Leukemia. Cancers. 2019; 11(12):1951. https://doi.org/10.3390/cancers11121951
Chicago/Turabian StylePadella, Antonella, Giorgia Simonetti, Giulia Paciello, George Giotopoulos, Carmen Baldazzi, Simona Righi, Martina Ghetti, Anna Stengel, Viviana Guadagnuolo, Rossella De Tommaso, and et al. 2019. "Novel and Rare Fusion Transcripts Involving Transcription Factors and Tumor Suppressor Genes in Acute Myeloid Leukemia" Cancers 11, no. 12: 1951. https://doi.org/10.3390/cancers11121951
APA StylePadella, A., Simonetti, G., Paciello, G., Giotopoulos, G., Baldazzi, C., Righi, S., Ghetti, M., Stengel, A., Guadagnuolo, V., De Tommaso, R., Papayannidis, C., Robustelli, V., Franchini, E., Ghelli Luserna di Rorà, A., Ferrari, A., Fontana, M. C., Bruno, S., Ottaviani, E., Soverini, S., ... Martinelli, G. (2019). Novel and Rare Fusion Transcripts Involving Transcription Factors and Tumor Suppressor Genes in Acute Myeloid Leukemia. Cancers, 11(12), 1951. https://doi.org/10.3390/cancers11121951