Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications

 ,
, 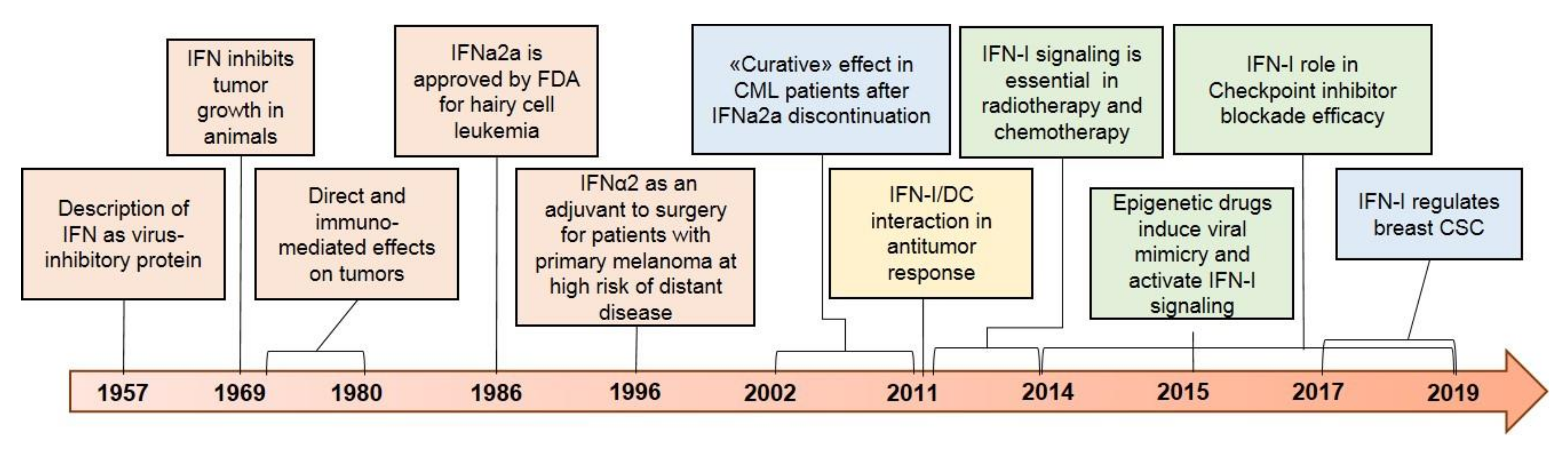{kind=link}
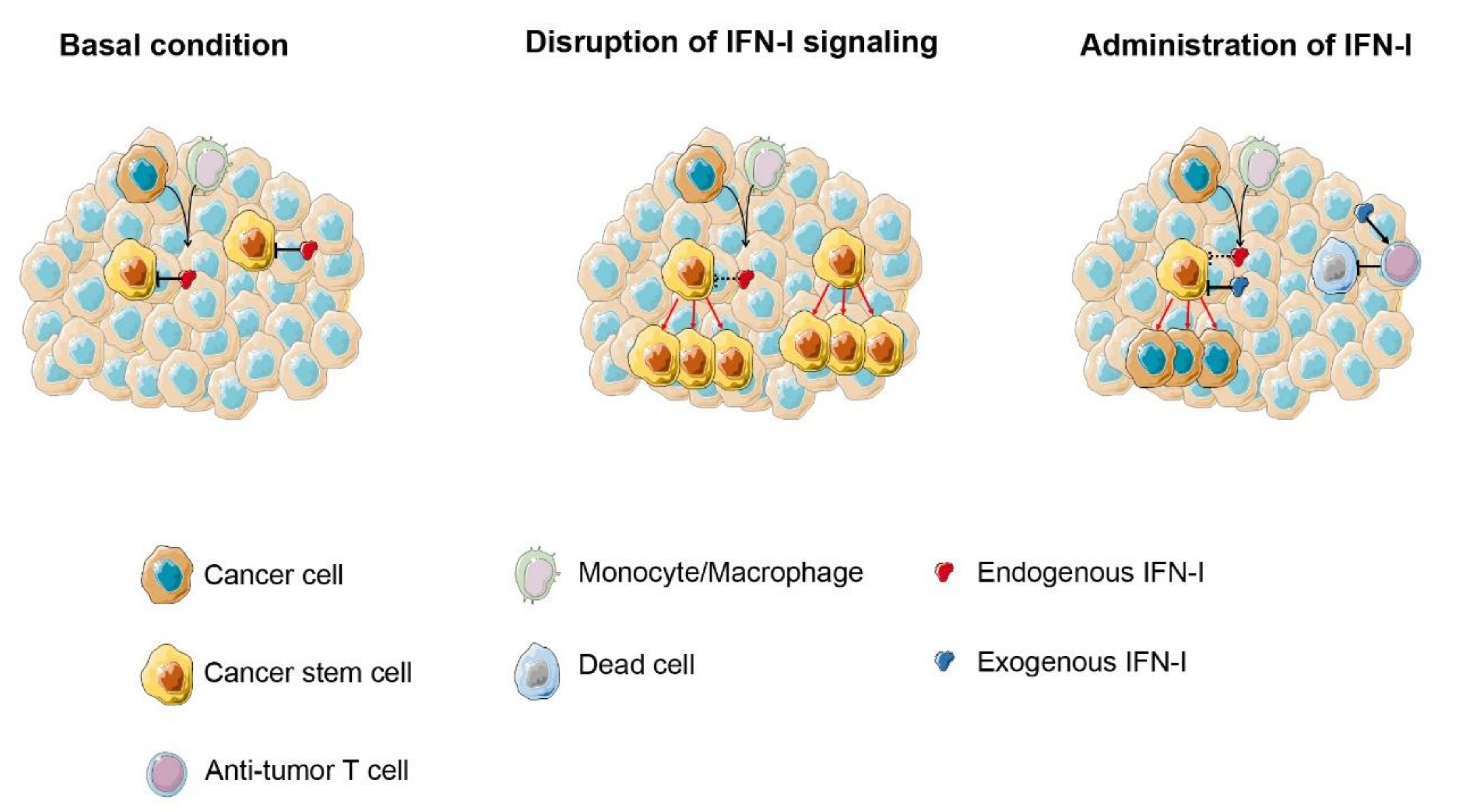{kind=link}
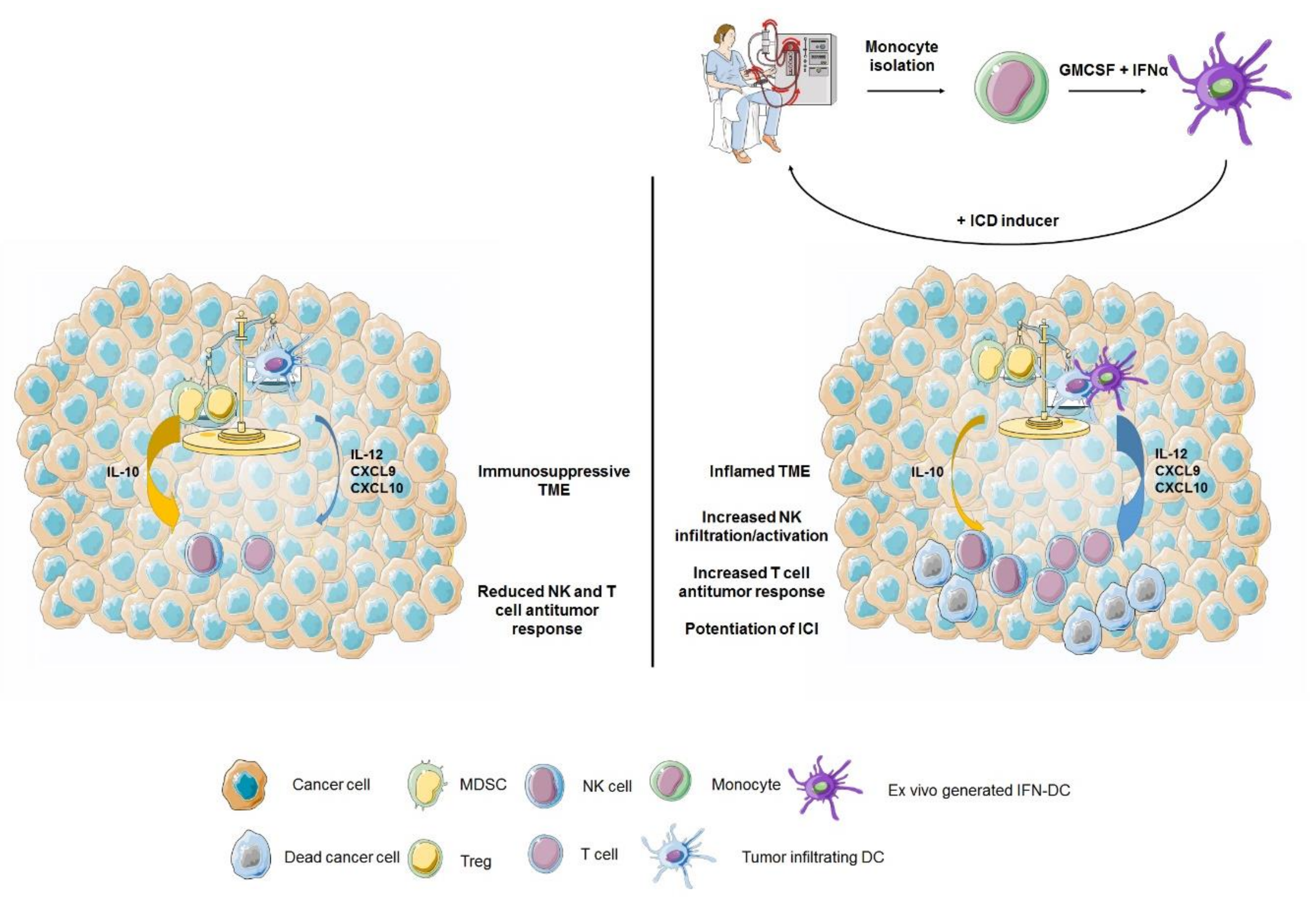{kind=link}
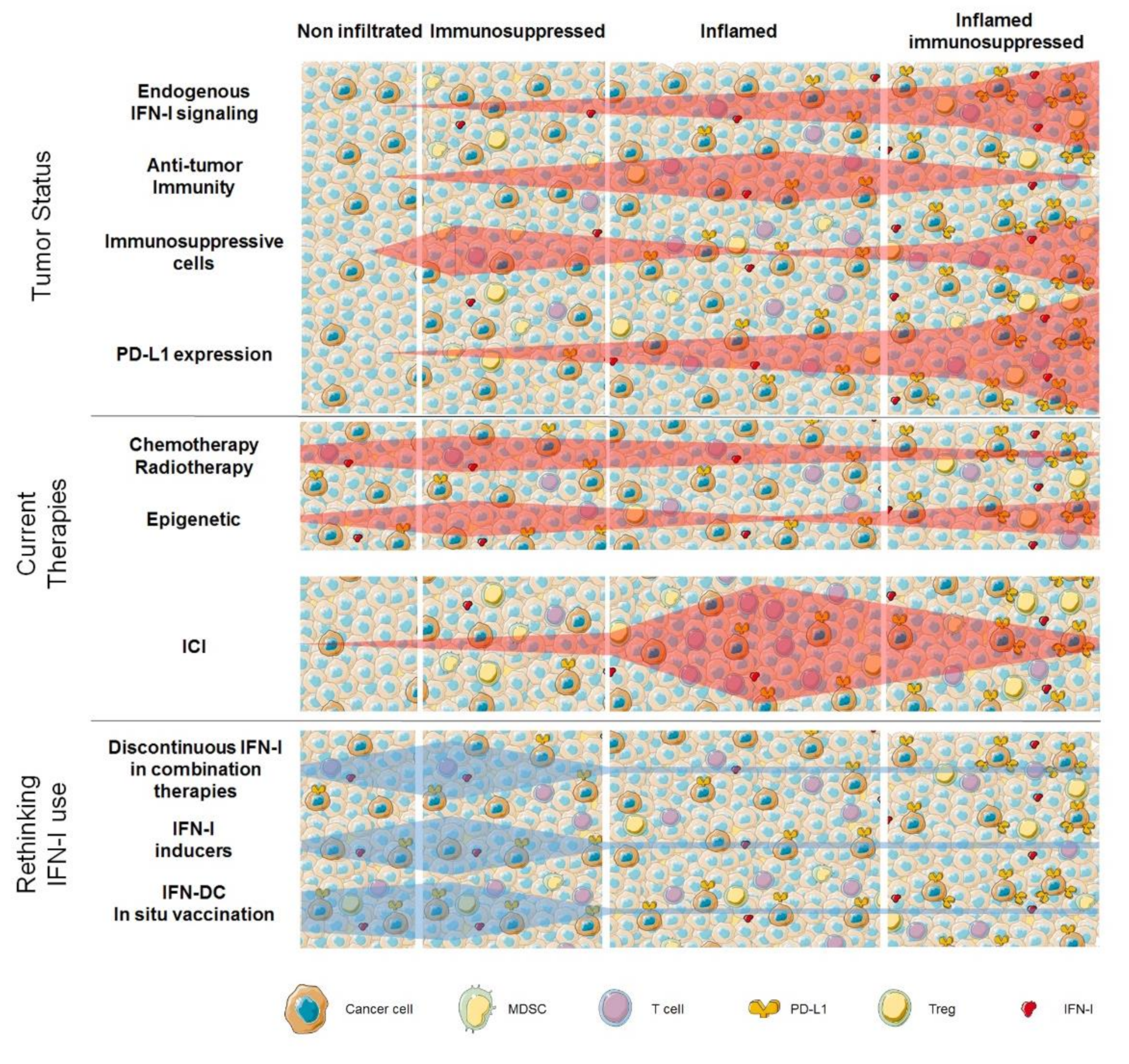{kind=link}
Abstract
1. Introduction
2. IFN-I in Combination Therapies
2.1. IFN-I and Chemo-Radiotherapy
2.2. IFN-I and ICI
2.3. IFN-I and Epigenetics
3. IFN-I in Antitumor Therapies Targeting Cancer Stem Cells (CSC)
4. IFN-I for Cancer Vaccination
4.1. IFN-I as Immune Adjuvant for Cancer Vaccines
4.2. IFN-α in DC-Based Combination Immunotherapy
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Gresser, I.; Bourali, C. Exogenous interferon and inducers of interferon in the treatment Balb-c mice inoculated with RC19 tumour cells. Nature 1969, 223, 844–845. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C. Interferons α and β in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Yang, J.C.; Atkins, M.B.; Disis, M.L. Toxicities of immunotherapy for the practitioner. J. Clin. Oncol. 2015, 33, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.F.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef]
- Castiello, L.; Sestili, P.; Schiavoni, G.; Dattilo, R.; Monque, D.M.; Ciaffoni, F.; Iezzi, M.; Lamolinara, A.; Sistigu, A.; Moschella, F.; et al. Disruption of IFN-I Signaling Promotes HER2/Neu Tumor Progression and Breast Cancer Stem Cells. Cancer Immunol. Res. 2018, 6, 658–670. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554. [Google Scholar] [CrossRef]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.G.; Liu, X.; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Katlinskaya, Y.V.; Katlinski, K.V.; Yu, Q.; Ortiz, A.; Daniel, P.; Brice, A.; Davar, D.; Sanders, C.; Kirkwood, J.M.; Xu, X.; et al. Suppression of type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep. 2016, 15, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; Agostinis, P.; Garg, A.D. Type I Interferons and Dendritic Cells in Cancer Immunotherapy, 1st ed.; Elsevier Inc.: Oxford, UK, 2019; pp. 217–262. [Google Scholar]
- Bracci, L.; Sistigu, A.; Proietti, E.; Moschella, F. The added value of type I interferons to cytotoxic treatments of cancer. Cytokine Growth Factor Rev. 2017, 36, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide Synergizes with Type I Interferons through Systemic Dendritic Cell Reactivation and Induction of Immunogenic Tumor Apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef]
- Palata, O.; Hradilova Podzimkova, N.; Nedvedova, E.; Umprecht, A.; Sadilkova, L.; Palova Jelinkova, L.; Spisek, R.; Adkins, I. Radiotherapy in Combination With Cytokine Treatment. Front. Oncol. 2019, 9, 367. [Google Scholar] [CrossRef]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef]
- Bald, T.; Landsberg, J.; Lopez-Ramos, D.; Renn, M.; Glodde, N.; Jansen, P.; Gaffal, E.; Steitz, J.; Tolba, R.; Kalinke, U.; et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014, 4, 674–687. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Goreczny, G.J.; Wilson, K.; Morrow, S.; DeCristo, M.J.; Ubellacker, J.M.; Qin, Y.; Laszewski, T.; Stover, D.G.; Barrera, V.; et al. Interferon Signaling is Diminished with Age and is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Wang, H.; Chauvin, J.M.; Pagliano, O.; Fourcade, J.J.; Ka, M.; Menna, C.; Rose, A.; Sander, C.; Borhani, A.A.; et al. Phase Ib/II study of pembrolizumab and pegylated-interferon alfa-2b in advanced melanoma. J. Clin. Oncol. 2018, 36, 3450–3458. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Hodi, F.S.; Thompson, J.A.; McDermott, D.F.; Hwu, W.-J.; Lawrence, D.P.; Dawson, N.A.; Wong, D.J.; Bhatia, S.; James, M.; et al. Pembrolizumab Plus Pegylated Interferon alfa-2b or Ipilimumab for Advanced Melanoma or Renal Cell Carcinoma: Dose-Finding Results from the Phase Ib KEYNOTE-029 Study. Clin. Cancer Res. 2018, 24, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S.; et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef]
- Liu, M.; Thomas, S.L.; DeWitt, A.K.; Zhou, W.; Madaj, Z.B.; Ohtani, H.; Baylin, S.B.; Liang, G.; Jones, P.A. Dual inhibition of DNA and histone methyltransferases increases viral mimicry in ovarian cancer cells. Cancer Res. 2018, 78, 5754–5760. [Google Scholar] [CrossRef]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.M.; Wang, T.L.; et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Fragale, A.; Romagnoli, G.; Licursi, V.; Buoncervello, M.; Del Vecchio, G.; Giuliani, C.; Parlato, S.; Leone, C.; De Angelis, M.; Canini, I.; et al. Antitumor Effects of Epidrug/IFNα Combination Driven by Modulated Gene Signatures in Both Colorectal Cancer and Dendritic Cells. Cancer Immunol. Res. 2017, 5, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X.; Delbrel, X.; Cony-Makhoul, P.; Fabères, C.; Boiron, J.M.; Barthe, C.; Bilhou-Nabéra, C.; Pigneux, A.; Marit, G.; Reiffers, J. Follow-Up of Complete Cytogenetic Remission in Patients With Chronic Myeloid Leukemia After Cessation of Interferon Alfa. J. Clin. Oncol. 2002, 20, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Veneri, D.; Tecchio, C.; De Matteis, G.; Paviati, E.; Benati, M.; Franchini, M.; Pizzolo, G. Long-term persistence of molecular response after discontinuation of interferon-alpha in two patients with chronic myeloid leukaemia. Blood Transfus. 2012, 10, 233–234. [Google Scholar] [PubMed]
- Talpaz, M.; Hehlmann, R.; Quintás-Cardama, A.; Mercer, J.; Cortes, J. Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia 2013, 27, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Latagliata, R.; Romano, A.; Mancini, M.; Breccia, M.; Carmosino, I.; Vozella, F.; Montagna, C.; Volpicelli, P.; De Angelis, F.; Petrucci, L.; et al. Discontinuation of alpha-interferon treatment in patients with chronic myeloid leukemia in long-lasting complete molecular response. Leuk. Lymphoma 2016, 57, 99–102. [Google Scholar] [CrossRef]
- Yokota, A.; Hirai, H.; Sato, R.; Adachi, H.; Sato, F.; Hayashi, Y.; Sato, A.; Kamio, N.; Miura, Y.; Nakano, M.; et al. C/EBPβ is a critical mediator of IFN-α-induced exhaustion of chronic myeloid leukemia stem cells. Blood Adv. 2019, 3, 476–488. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Liu, D.D.; Choudhury, A.; Hang, X.; Wei, Y.; Zamalloa, J.; Alfaro-Aco, R.; Chakrabarti, R.; Jiang, Y.Z.; Koh, B.I.; et al. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a–LCOR axis. Nat. Cell Biol. 2017, 19, 711–723. [Google Scholar] [CrossRef]
- Doherty, M.R.; Cheon, H.; Junk, D.J.; Vinayak, S.; Varadan, V.; Telli, M.L.; Ford, J.M.; Stark, G.R.; Jackson, M.W. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13792–13797. [Google Scholar] [CrossRef]
- Buoncervello, M.; Romagnoli, G.; Buccarelli, M.; Fragale, A.; Toschi, E.; Parlato, S.; Lucchetti, D.; Macchia, D.; Spada, M.; Canini, I.; et al. IFN-α potentiates the direct and immune-mediated antitumor effects of epigenetic drugs on both metastatic and stem cells of colorectal cancer. Oncotarget 2016, 7, 26361–26373. [Google Scholar] [CrossRef]
- Doherty, M.R.; Jackson, M.W. The Critical, Clinical Role of Interferon-Beta in Regulating Cancer Stem Cell Properties in Triple-Negative Breast Cancer. DNA Cell Biol. 2018, 37, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Rizza, P.; Moretti, F.; Capone, I.; Belardelli, F. Role of type I interferon in inducing a protective immune response: Perspectives for clinical applications. Cytokine Growth Factor Rev. 2015, 26, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Miquilena-Colina, M.E.; Lozano-Rodríguez, T.; García-Pozo, L.; Sáez, A.; Rizza, P.; Capone, I.; Rapicetta, M.; Chionne, P.; Capobianchi, M.; Selleri, M.; et al. Recombinant interferon-alpha2b improves immune response to hepatitis B vaccination in haemodialysis patients: Results of a randomised clinical trial. Vaccine 2009, 27, 5654–5660. [Google Scholar] [CrossRef] [PubMed]
- Di Pucchio, T.; Pilla, L.; Capone, I.; Ferrantini, M.; Montefiore, E.; Urbani, F.; Patuzzo, R.; Pennacchioli, E.; Santinami, M.; Cova, A.; et al. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gplOO peptides plus IFN-α results in the activation of specific CD8+ T cells and monocyte/dendritic cell precursors. Cancer Res. 2006, 66, 4943–4951. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8 + T cell responses through CD8α + dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef]
- Santini, S.M.; Lapenta, C.; Logozzi, M.; Parlato, S.; Spada, M.; Di Pucchio, T.; Belardelli, F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 2000, 191, 1777–1788. [Google Scholar] [CrossRef]
- Santodonato, L.; D’Agostino, G.; Nisini, R.; Mariotti, S.; Monque, D.M.; Spada, M.; Lattanzi, L.; Perrone, M.P.; Andreotti, M.; Belardelli, F.; et al. Monocyte-derived dendritic cells generated after a short-term culture with IFN-alpha and granulocyte-macrophage colony-stimulating factor stimulate a potent Epstein-Barr virus-specific CD8+ T cell response. J. Immunol. 2003, 170, 5195–5202. [Google Scholar] [CrossRef]
- Spadaro, F.; Lapenta, C.; Donati, S.; Abalsamo, L.; Barnaba, V.; Belardelli, F.; Santini, S.M.; Ferrantini, M. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood 2012, 119, 1407–1417. [Google Scholar] [CrossRef]
- Lapenta, C.; Donati, S.; Spadaro, F.; Castaldo, P.; Belardelli, F.; Cox, M.C.; Santini, S.M. NK Cell Activation in the Antitumor Response Induced by IFN-α Dendritic Cells Loaded with Apoptotic Cells from Follicular Lymphoma Patients. J. Immunol. 2016, 197, 795–806. [Google Scholar] [CrossRef]
- Rozera, C.; Cappellini, G.A.; D’Agostino, G.; Santodonato, L.; Castiello, L.; Urbani, F.; Macchia, I.; Aricò, E.; Casorelli, I.; Sestili, P.; et al. Intratumoral injection of IFN-alpha dendritic cells after dacarbazine activates anti-tumor immunity: Results from a phase I trial in advanced melanoma. J. Transl. Med. 2015, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.C.; Castiello, L.; Mattei, M.; Santodonato, L.; D’Agostino, G.; Muraro, E.; Martorelli, D.; Lapenta, C.; Di Napoli, A.; Di Landro, F.; et al. Clinical and antitumor immune responses In Relapsed/Refractory Follicular Lymphoma patients after intranodal injections of IFNα-Dendritic Cells and Rituximab. Clin. Cancer Res. 2019, 8, 1557. [Google Scholar] [CrossRef]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Santini, S.M.; Lapenta, C.; Donati, S.; Spadaro, F.; Belardelli, F.; Ferrantini, M. Interferon-α-Conditioned Human Monocytes Combine a Th1-Orienting Attitude with the Induction of Autologous Th17 Responses: Role of IL-23 and IL-12. PLoS ONE 2011, 6, e17364. [Google Scholar] [CrossRef]
- Castiello, L.; Aricò, E.; D’Agostino, G.; Santodonato, L.; Belardelli, F. In situ Vaccination by Direct Dendritic Cell Inoculation: The Coming of Age of an Old Idea? Front. Immunol. 2019, 10, 2303. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Gracia, J.L.P.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef]
- Cauwels, A.; Van Lint, S.; Paul, F.; Garcin, G.; De Koker, S.; Van Parys, A.; Wueest, T.; Gerlo, S.; Van der Heyden, J.; Bordat, Y.; et al. Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments. Cancer Res. 2018, 78, 463–474. [Google Scholar] [CrossRef]
- Cauwels, A.; Van Lint, S.; Garcin, G.; Bultinck, J.; Paul, F.; Gerlo, S.; Van der Heyden, J.; Bordat, Y.; Catteeuw, D.; De Cauwer, L.; et al. A safe and highly efficient tumor-targeted type I interferon immunotherapy depends on the tumor microenvironment. Oncoimmunology 2018, 7, e1398876. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers 2019, 11, 1943. https://doi.org/10.3390/cancers11121943
Aricò E, Castiello L, Capone I, Gabriele L, Belardelli F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers. 2019; 11(12):1943. https://doi.org/10.3390/cancers11121943
Chicago/Turabian StyleAricò, Eleonora, Luciano Castiello, Imerio Capone, Lucia Gabriele, and Filippo Belardelli. 2019. "Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications" Cancers 11, no. 12: 1943. https://doi.org/10.3390/cancers11121943
APA StyleAricò, E., Castiello, L., Capone, I., Gabriele, L., & Belardelli, F. (2019). Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers, 11(12), 1943. https://doi.org/10.3390/cancers11121943