Cancer Is Associated with Alterations in the Three-Dimensional Organization of the Genome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. A Total of 1467 Topologically Associating Domains (TADs) Constitute the Consensus TAD Map of the Human Genome
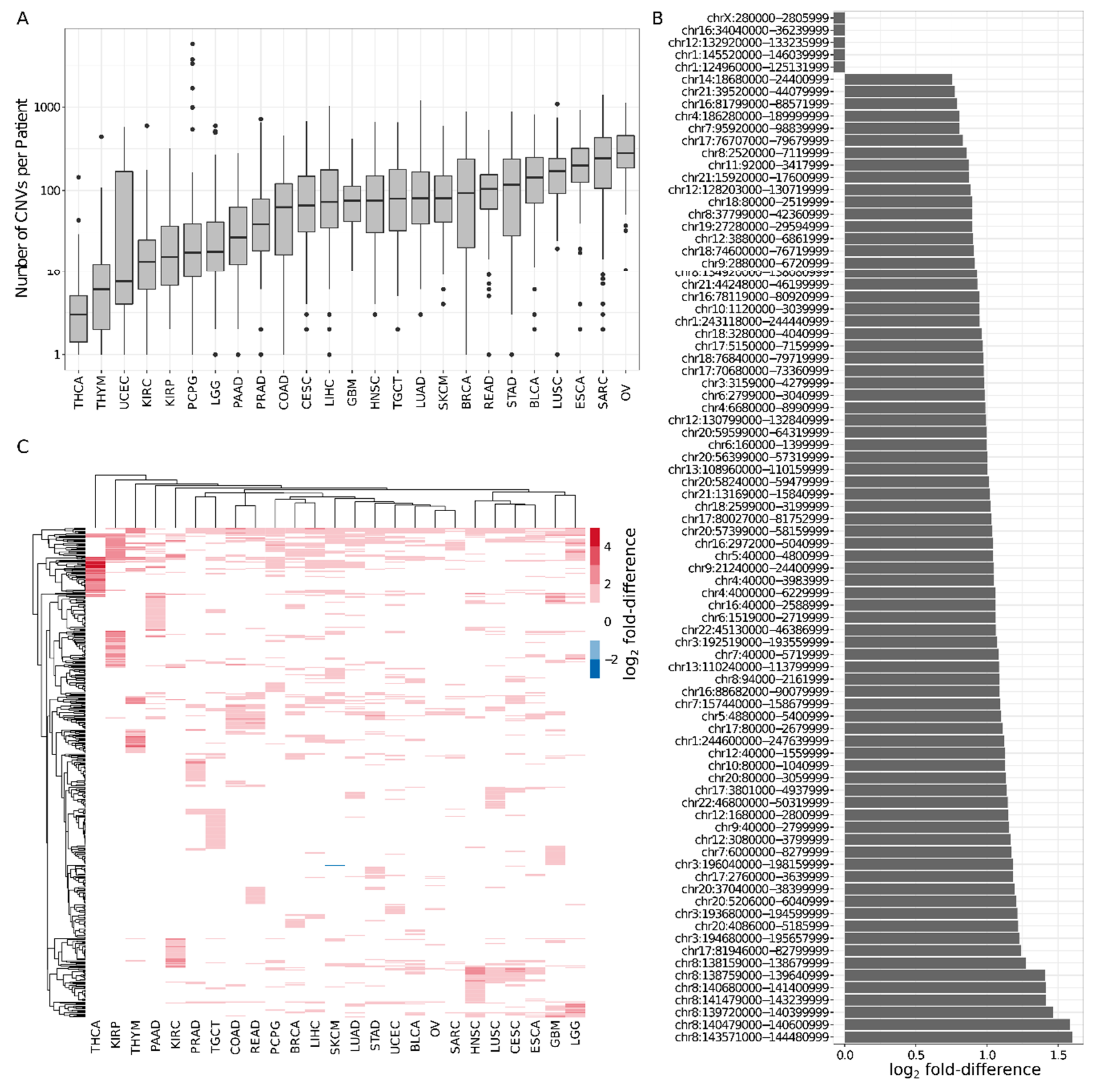
2.2. Six Percent of Consensus TADs Are Enriched or Depleted for Cancer-Related CNVs
2.3. TADs Enriched for CNVs Are Valuable Prognostic Biomarkers in Cancer
2.4. Thirty-Four Percent of Prognostic TADs Tend to Undergo Large Structural Changes in Cancer
3. Discussion
4. Materials and Methods
4.1. Topologically Associating Domain (TAD) Maps
4.2. TAD Size Comparison Between Normal and Cancer States
4.3. Similarity Between TAD Maps of Different Tissues/Cell Lines
4.4. Construction of Consensus TADs
4.4.1. Contribution of Each Tissue/Cell Line in Total Gene Expression Divergence
4.4.2. Conservation Scores
4.5. Constitutive and Perturbed TADs
4.6. Enrichment Analysis of CTCF Peaks and Housekeeping Genes (HK Genes) in Consensus TADs and TBRs
4.7. Cancer-Related Copy Number Variants (CNVs)
4.8. TADs Enriched/Depleted for CNVs
4.9. Pan-Cancer Genes
4.10. Functional and Pathway Analysis
4.11. Survival Analysis
4.11.1. TAD- and Pan-Cancer Gene-Based Overall Survival Cox Regression Models
4.11.2. Prognostic TADs and Pan-Cancer Genes
4.11.3. Patient Stratification
4.12. CNV Densities in Constitutive and Perturbed TADs
4.13. Expression Levels of NOCR2 in SARC Patients
4.14. Independent Validation of the OV TAD-Based Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.H.; Corces, V.G. A CTCF Code for 3D Genome Architecture. Cell 2015, 162, 703–705. [Google Scholar] [CrossRef]
- Acemel, R.D.; Maeso, I.; Gomez-Skarmeta, J.L. Topologically associated domains: A successful scaffold for the evolution of gene regulation in animals. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e265. [Google Scholar] [CrossRef]
- Galupa, R.; Heard, E. Topologically Associating Domains in Chromosome Architecture and Gene Regulatory Landscapes during Development, Disease, and Evolution. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; Van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Battulin, N.; Fishman, V.S.; Mazur, A.M.; Pomaznoy, M.; Khabarova, A.A.; Afonnikov, D.A.; Prokhortchouk, E.B.; Serov, O.L. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015, 16, 77. [Google Scholar] [CrossRef]
- Valton, A.L.; Dekker, J. TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 2016, 36, 34–40. [Google Scholar] [CrossRef]
- Kaiser, V.B.; Semple, C.A. When TADs go bad: Chromatin structure and nuclear organisation in human disease. F1000 Research 2017, 6. [Google Scholar] [CrossRef]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nature Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; You, J.; Huang, T.; Cai, Y.D. Classification of non-small cell lung cancer based on copy number alterations. PLoS ONE 2014, 9, e88300. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, M.; Zhang, P.; Huang, T. Classification of cancers based on copy number variation landscapes. Biochim. Biophys. Acta 2016, 1860, 2750–2755. [Google Scholar] [CrossRef] [PubMed]
- Klopocki, E.; Mundlos, S. Copy-number variations, noncoding sequences, and human phenotypes. Annu. Rev. Genomics Hum. Genet. 2011, 12, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, M.; Mundlos, S. Looking beyond the genes: The role of non-coding variants in human disease. Hum. Mol. Genet. 2016, 25, R157–R165. [Google Scholar] [CrossRef]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schopflin, R.; Kraft, K.; Kempfer, R.; Jerkovic, I.; Chan, W.L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Muller, L.; Aigner, P.; Stoiber, D. Type I Interferons and Natural Killer Cell Regulation in Cancer. Front. Immunol. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.Q.; Webber, C.; Ponting, C.P. Bias of selection on human copy-number variants. PLoS Genet. 2006, 2, e20. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.M.; Guo, C.; Zhang, J. Nuclear receptor corepressor complexes in cancer: Mechanism, function and regulation. Am. J. Clin. Exp. Urol. 2014, 2, 169–187. [Google Scholar] [PubMed]
- Phelps, M.P.; Bailey, J.N.; Vleeshouwer-Neumann, T.; Chen, E.Y. CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 15090–15095. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.S.; Leavy, O.C.; Dryden, N.H.; Maguire, S.; Johnson, N.; Fedele, V.; Simigdala, N.; Martin, L.A.; Andrews, S.; Wingett, S.W.; et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat. Commun. 2018, 9, 1028. [Google Scholar] [CrossRef] [PubMed]
- Golloshi, R.; Sanders, J.T.; McCord, R.P. Iteratively improving Hi-C experiments one step at a time. Methods 2018, 142, 47–58. [Google Scholar] [CrossRef]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal-a one-stop shop for cancer genomics data. Database J. Biol. Databases Curation 2011, 2011, bar026. [Google Scholar] [CrossRef]
- D’Angelo, E.; Prat, J. Uterine sarcomas: A review. Gynecol. Oncol. 2010, 116, 131–139. [Google Scholar] [CrossRef]
- Tsuyoshi, H.; Yoshida, Y. Molecular biomarkers for uterine leiomyosarcoma and endometrial stromal sarcoma. Cancer Sci. 2018, 109, 1743–1752. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965. [Google Scholar] [CrossRef] [PubMed]
- Kloetgen, A.; Thandapani, P.; Ntziachristos, P.; Ghebrechristos, Y.; Nomikou, S.; Lazaris, C.; Chen, X.; Hu, H.; Bakogianni, S.; Wang, J.; et al. Dynamic 3D chromosomal landscapes in acute leukemia. bioRxiv 2019, 10. [Google Scholar] [CrossRef]
- Diaz, N.; Kruse, K.; Erdmann, T.; Staiger, A.M.; Ott, G.; Lenz, G.; Vaquerizas, J.M. Chromatin conformation analysis of primary patient tissue using a low input Hi-C method. Nat. Commun. 2018, 9, 4938. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef]
- Leung, D.; Jung, I.; Rajagopal, N.; Schmitt, A.; Selvaraj, S.; Lee, A.Y.; Yen, C.A.; Lin, S.; Lin, Y.; Qiu, Y.; et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature 2015, 518, 350–354. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Stone, E.A.; Sidow, A. Constructing a meaningful evolutionary average at the phylogenetic center of mass. BMC Bioinform. 2007, 8, 222. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucl. Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Harrell, F.E., Jr.; Califf, R.M.; Pryor, D.B.; Lee, K.L.; Rosati, R.A. Evaluating the yield of medical tests. JAMA 1982, 247, 2543–2546. [Google Scholar] [CrossRef]
- Laimighofer, M.; Krumsiek, J.; Buettner, F.; Theis, F.J. Unbiased Prediction and Feature Selection in High-Dimensional Survival Regression. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2016, 23, 279–290. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in S. Available online: https://www.mayo.edu/research/documents/tr53pdf/doc-10027379 (accessed on 28 August 2019).
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Barth, N.K.H.; Pilarsky, C.; Taher, L. Cancer Is Associated with Alterations in the Three-Dimensional Organization of the Genome. Cancers 2019, 11, 1886. https://doi.org/10.3390/cancers11121886
Li L, Barth NKH, Pilarsky C, Taher L. Cancer Is Associated with Alterations in the Three-Dimensional Organization of the Genome. Cancers. 2019; 11(12):1886. https://doi.org/10.3390/cancers11121886
Chicago/Turabian StyleLi, Lifei, Nicolai K. H. Barth, Christian Pilarsky, and Leila Taher. 2019. "Cancer Is Associated with Alterations in the Three-Dimensional Organization of the Genome" Cancers 11, no. 12: 1886. https://doi.org/10.3390/cancers11121886
APA StyleLi, L., Barth, N. K. H., Pilarsky, C., & Taher, L. (2019). Cancer Is Associated with Alterations in the Three-Dimensional Organization of the Genome. Cancers, 11(12), 1886. https://doi.org/10.3390/cancers11121886