HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
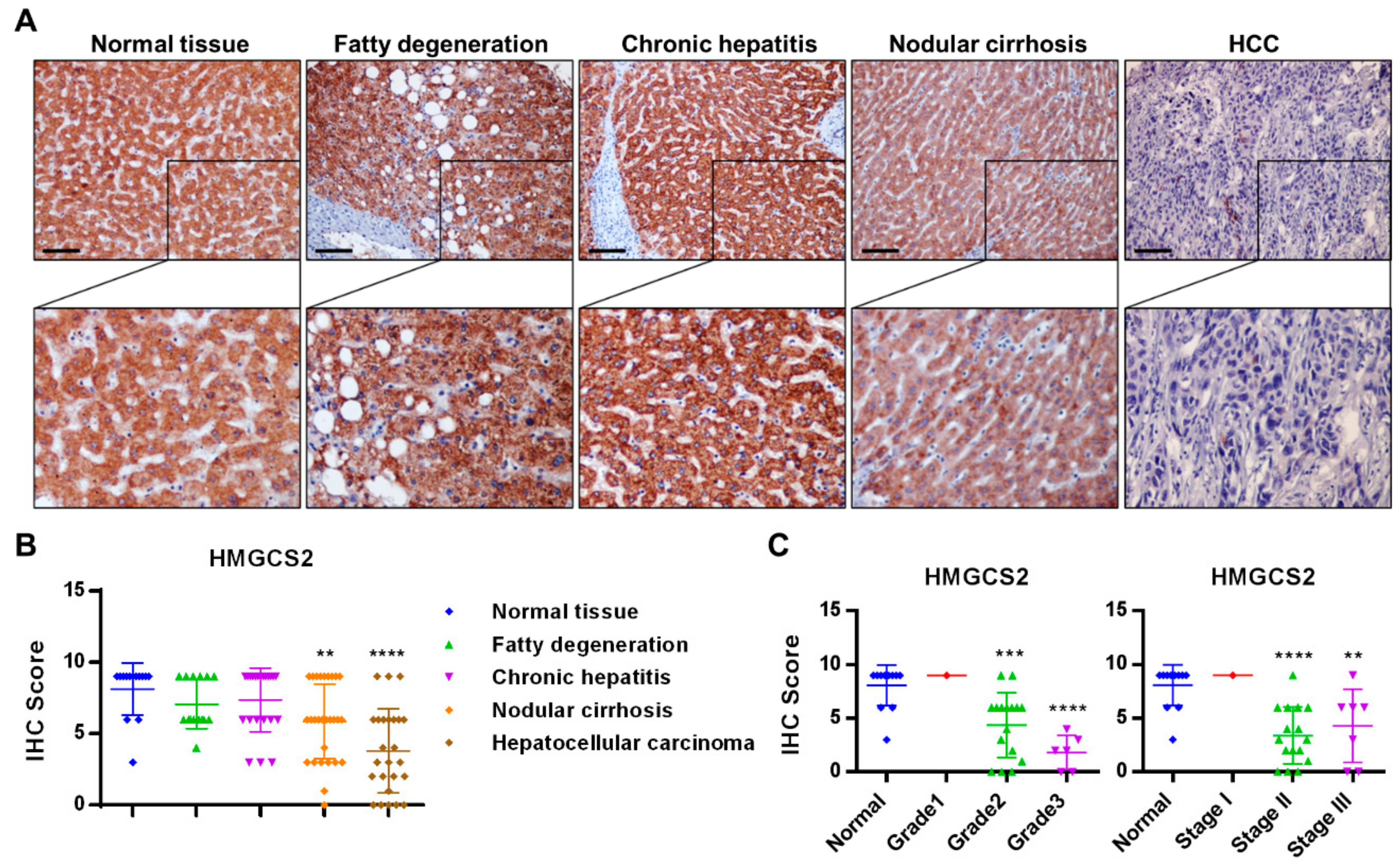
2.1. HMGCS2 Expression Is Decreased in Liver Cancer Tissues
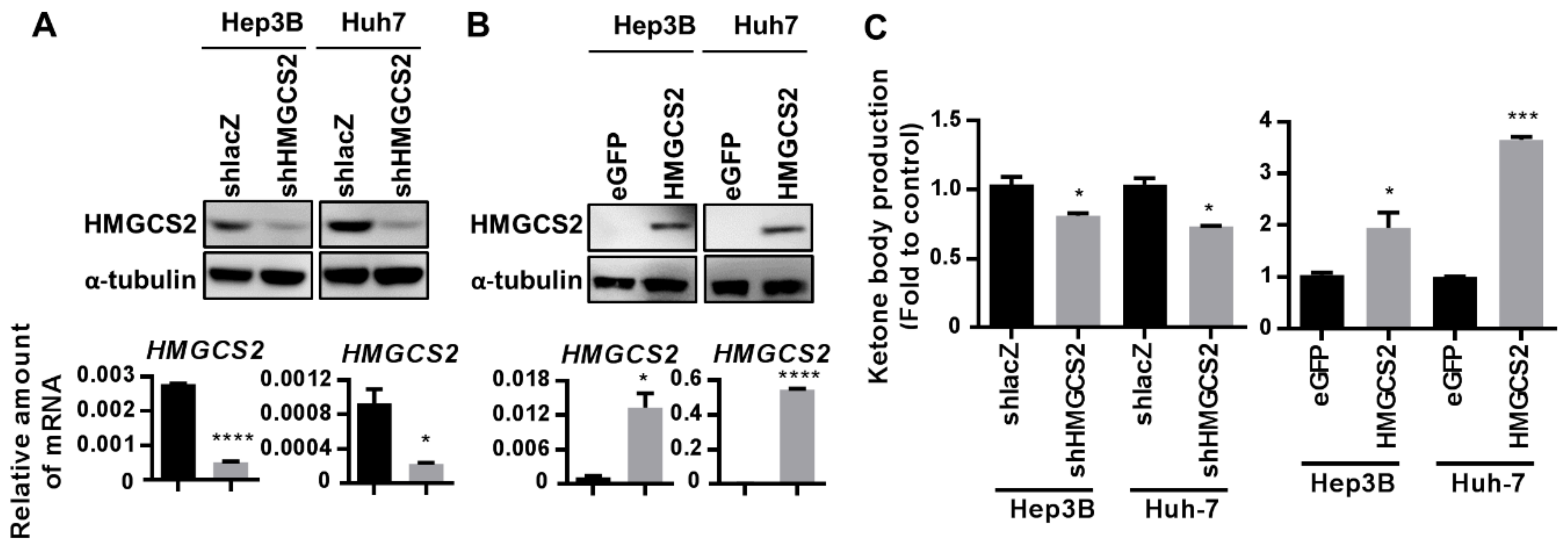
2.2. Establishment of Cell Lines with Stable HMGCS2 Overexpression and Knockdown
2.3. Genes and Biological Functions Affected by Downregulated Expression of HMGCS2 in HCC Cells
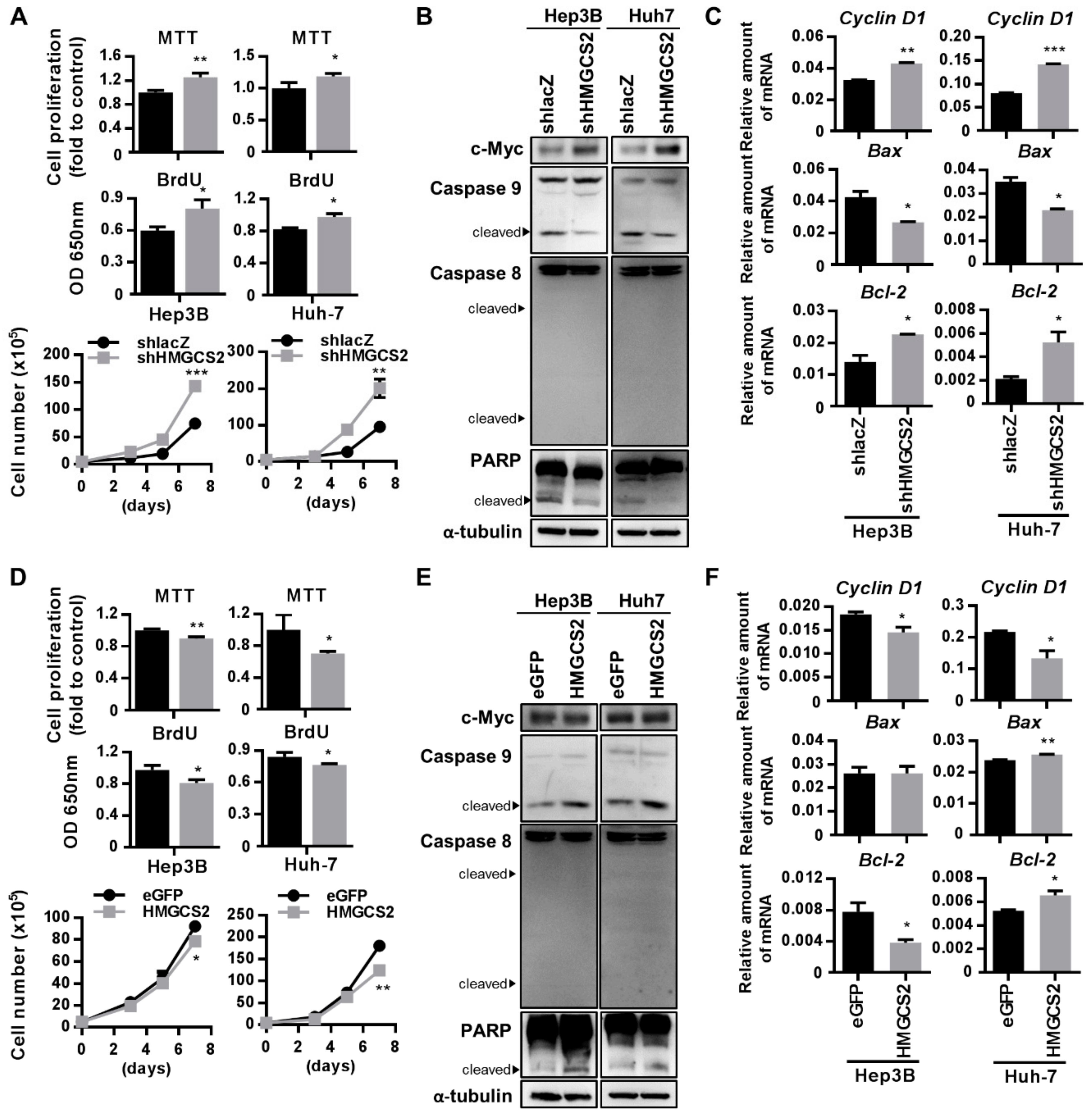
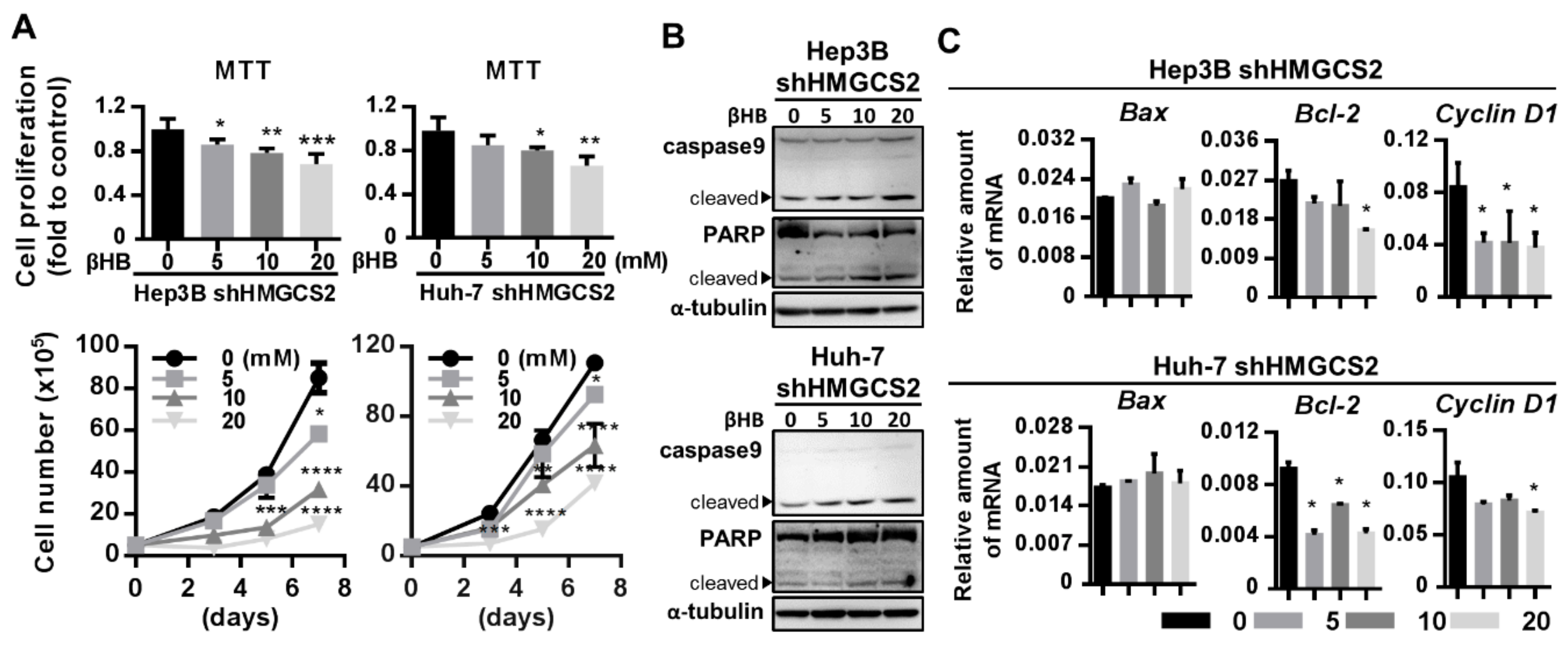
2.4. HMGCS2 Suppresses Liver Cancer Cell Growth by Increasing Apoptosis and Inhibiting Proliferation
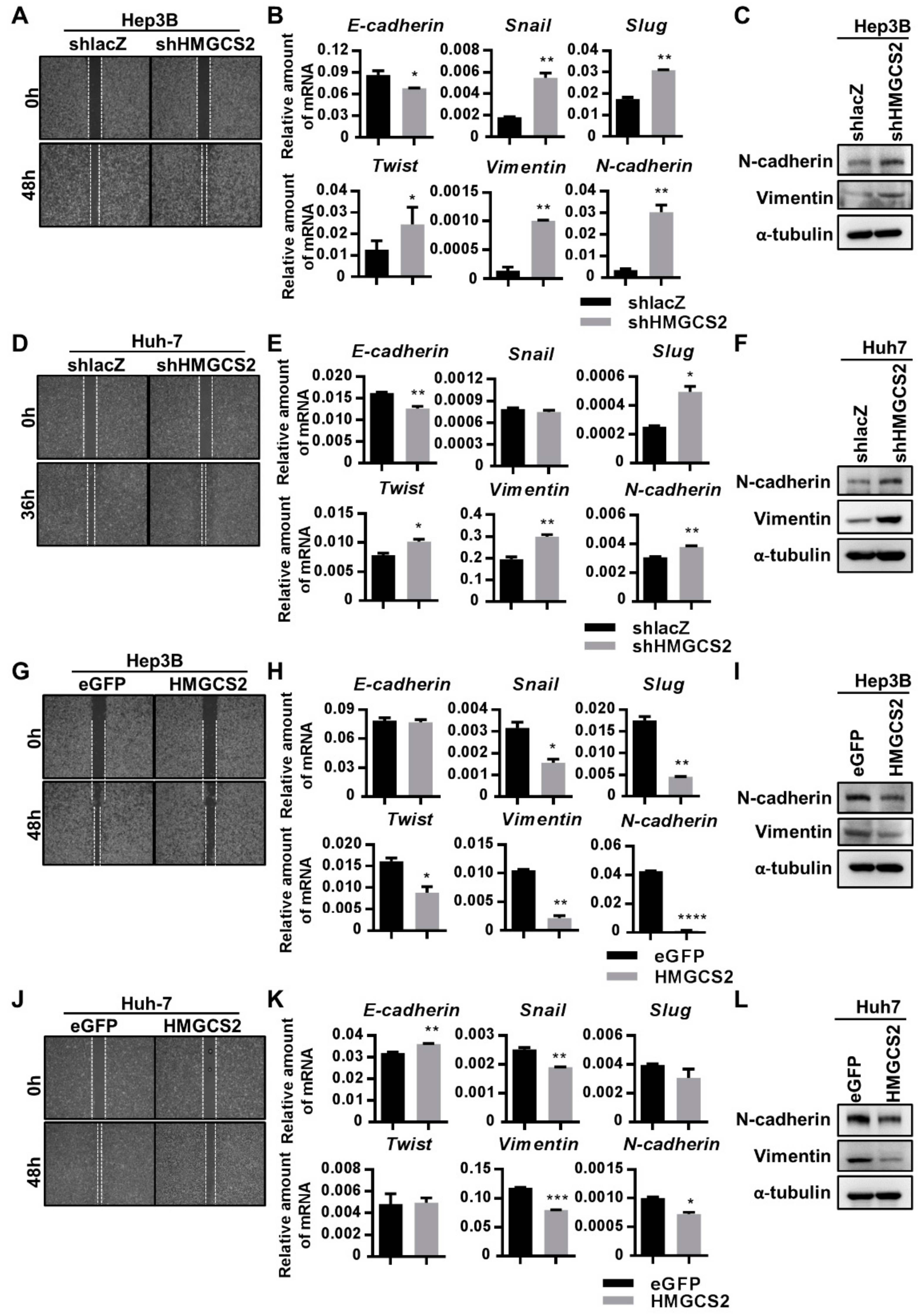
2.5. Low Expression of HMGCS2 Is Correlated with Enhanced Migration Ability
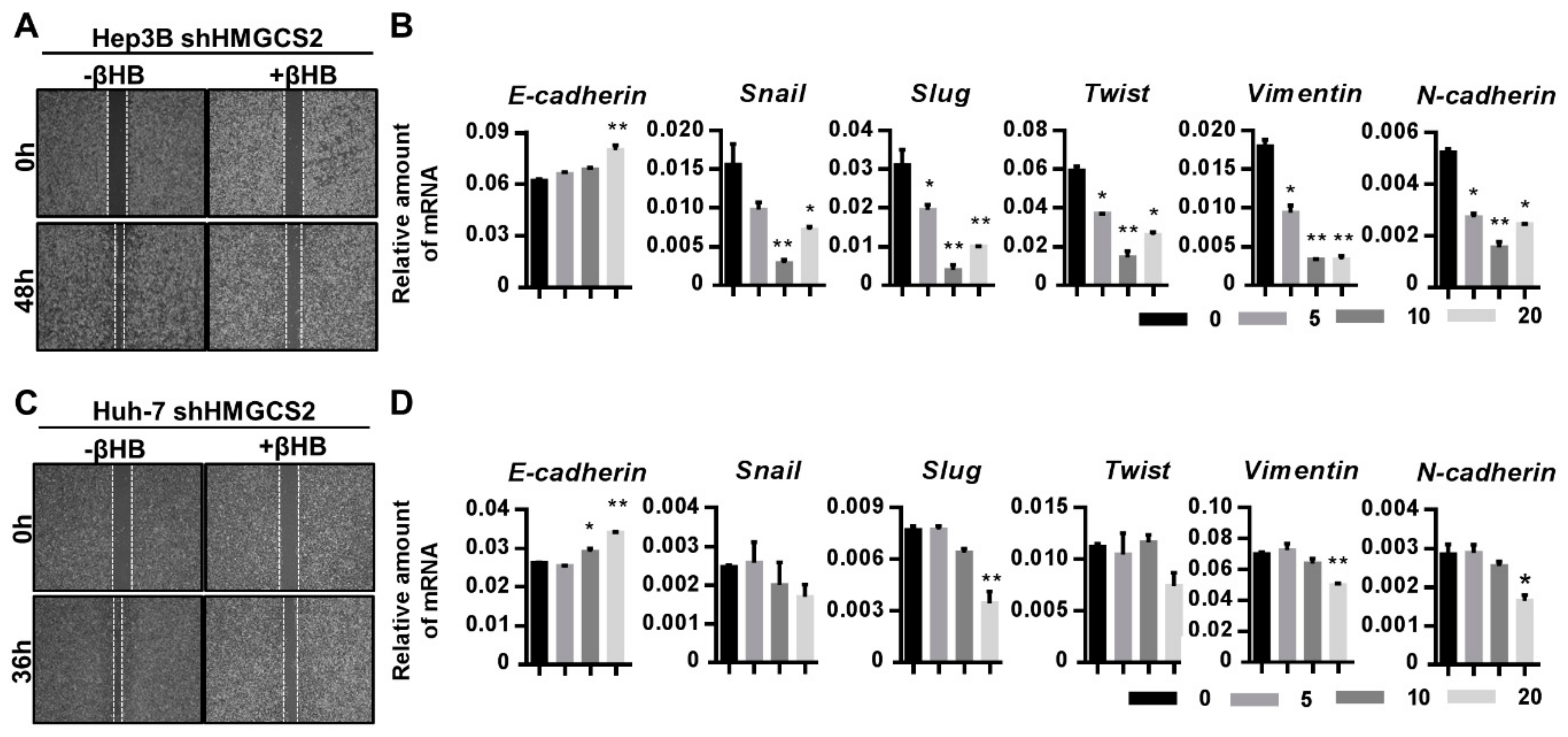
2.6. Ketone Body Supplementation Reduces the Increase in the Proliferation and Migration Abilities of HMGCS2 Knockdown Cells
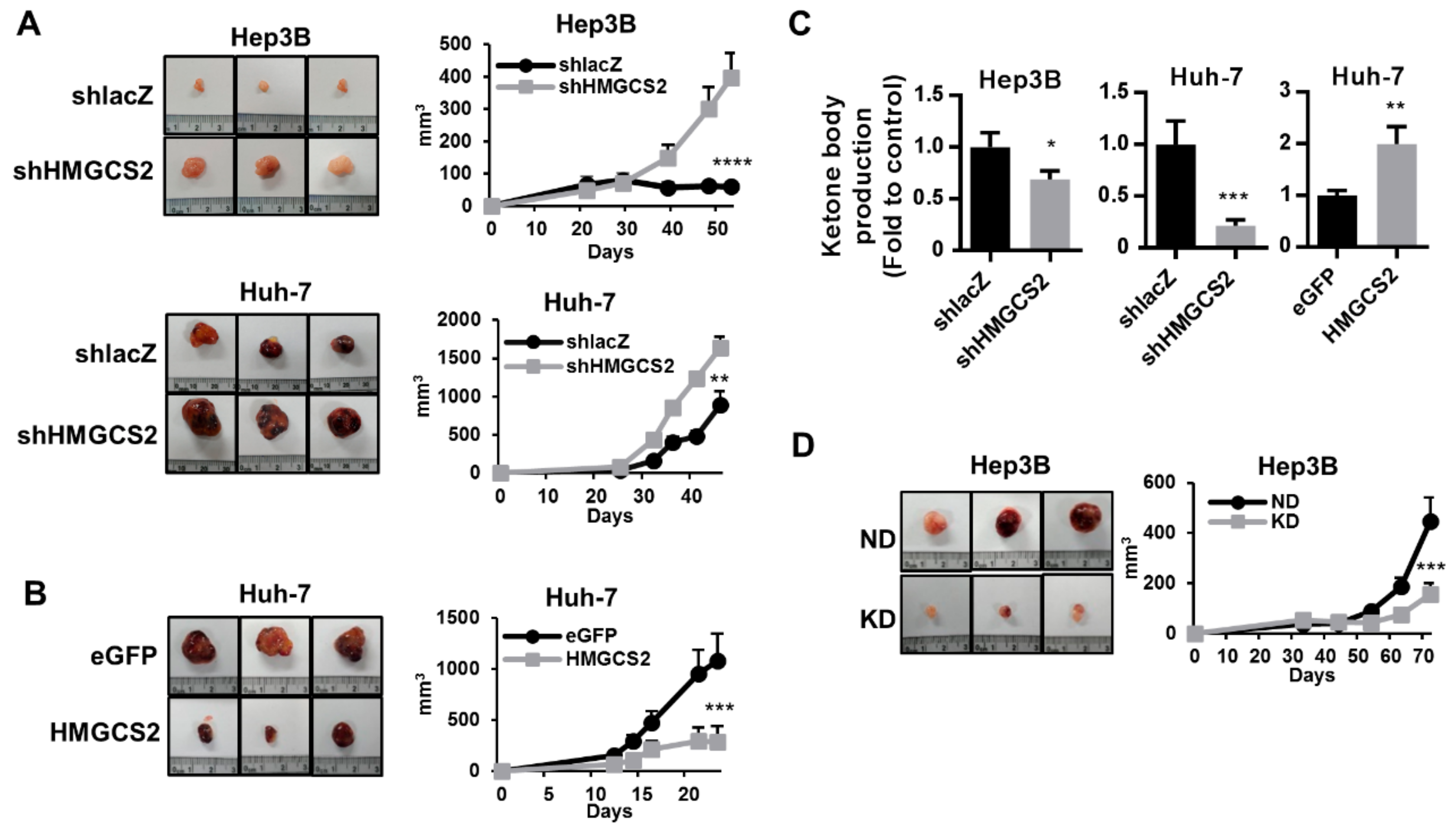
2.7. HMGCS2-Related Ketone Production Affects HCC Tumor Growth In Vivo
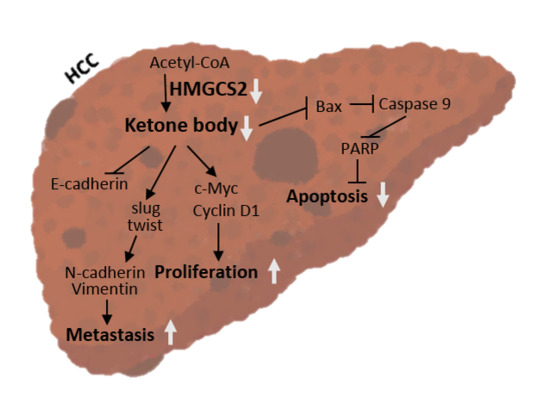
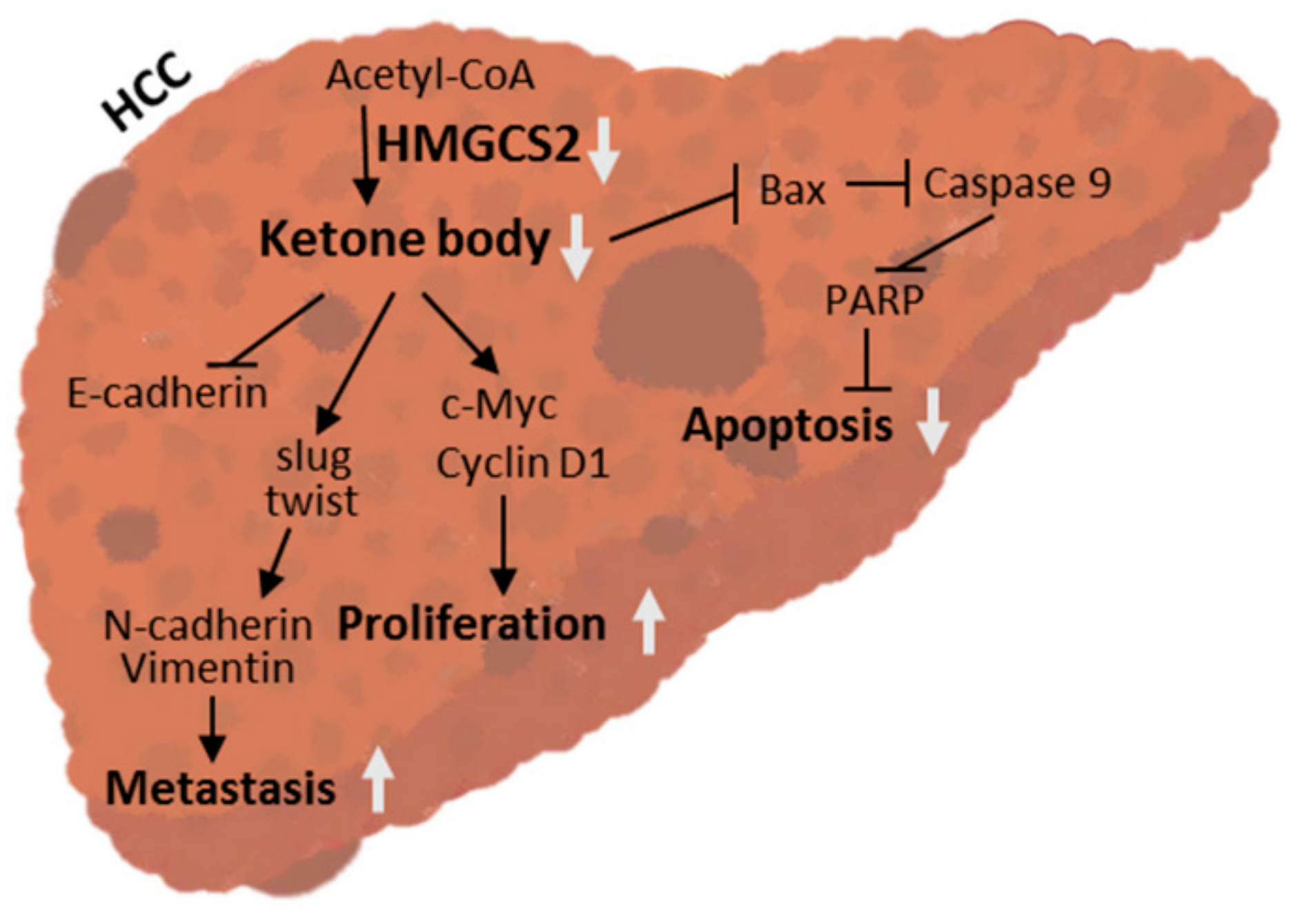
3. Discussion
4. Materials and Methods
4.1. Immunohistochemical Staining
4.2. Cell Culture, Plasmids, and Lentivirus Infection
4.3. Colorimetric β-Hydroxybutyrate Assay
4.4. Trypan Blue Cell Viability Assay
4.5. MTT Cell Viability Assay
4.6. BrdU Cell Proliferation Assay
4.7. Wound-Healing Migration Assay
4.8. Western Blot Analysis
4.9. Real-Time PCR (QPCR)
4.10. Subcutaneous Xenograft Model
4.11. β-Hydroxybutyrate Treatment
4.12. Gene Expression Profiling
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.; Lee, H.; Park, J.; Kim, S.H.; Park, J. Targeting cancer metabolism—Revisiting the warburg effects. Toxicol. Res. 2016, 32, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, D.M.; Namboodiri, A.M.; Moffett, J.R. Acetate as a metabolic and epigenetic modifier of cancer therapy. J. Cell Biochem. 2016, 117, 574–588. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- US Cancer Statistics Working Group. United States Cancer Statistics: 1999–2006 Incidence and Mortality Web-Based Report; US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute: Atlanta, GA, USA, 2010.
- Forner, A.; Gilabert, M.; Bruix, J.; Raoul, J.L. Treatment of intermediate-stage hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2014, 11, 525–535. [Google Scholar] [CrossRef]
- Zarrinpar, A. Metabolic pathway inhibition in liver cancer. SLAS Technol. 2017, 22, 237–244. [Google Scholar] [CrossRef][Green Version]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef]
- Mello, T.; Materozzi, M.; Galli, A. Ppars and mitochondrial metabolism: From nafld to hcc. PPAR Res. 2016, 2016, 7403230. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Ketone body utilization drives tumor growth and metastasis. Cell Cycle 2012, 11, 3964–3971. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef]
- Zhang, W.W.; Churchill, S.; Lindahl, R.; Churchill, P. Regulation of d-beta-hydroxybutyrate dehydrogenase in rat hepatoma cell lines. Cancer Res. 1989, 49, 2433–2437. [Google Scholar]
- Grabacka, M.M.; Wilk, A.; Antonczyk, A.; Banks, P.; Walczyk-Tytko, E.; Dean, M.; Pierzchalska, M.; Reiss, K. Fenofibrate induces ketone body production in melanoma and glioblastoma cells. Front. Endocrinol. 2016, 7, 5. [Google Scholar] [CrossRef]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [Google Scholar] [CrossRef]
- Poff, A.M.; Ari, C.; Arnold, P.; Seyfried, T.N.; D’Agostino, D.P. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int. J. Cancer 2014, 135, 1711–1720. [Google Scholar] [CrossRef]
- Shafqat, N.; Turnbull, A.; Zschocke, J.; Oppermann, U.; Yue, W.W. Crystal structures of human hmg-coa synthase isoforms provide insights into inherited ketogenesis disorders and inhibitor design. J. Mol. Biol. 2010, 398, 497–506. [Google Scholar] [CrossRef]
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772. [Google Scholar]
- Konig, B.; Fischer, S.; Schlotte, S.; Wen, G.; Eder, K.; Stangl, G.I. Monocarboxylate transporter 1 and cd147 are up-regulated by natural and synthetic peroxisome proliferator-activated receptor alpha agonists in livers of rodents and pigs. Mol. Nutr. Food Res. 2010, 54, 1248–1256. [Google Scholar] [CrossRef]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of pparalpha. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Mascaro, C.; Buesa, C.; Ortiz, J.A.; Haro, D.; Hegardt, F.G. Molecular cloning and tissue expression of human mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase. Arch. Biochem. Biophys. 1995, 317, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shidfar, A.; Ivancic, D.; Ranjan, M.; Liu, L.; Choi, M.R.; Parimi, V.; Gursel, D.B.; Sullivan, M.E.; Najor, M.S.; et al. Overexpression of lipid metabolism genes and pbx1 in the contralateral breasts of women with estrogen receptor-negative breast cancer. Int. J. Cancer 2017, 140, 2484–2497. [Google Scholar] [CrossRef] [PubMed]
- Gromov, P.; Espinoza, J.A.; Talman, M.L.; Honma, N.; Kroman, N.; Timmermans Wielenga, V.; Moreira, J.M.; Gromova, I. Fabp7 and hmgcs2 are novel protein markers for apocrine differentiation categorizing apocrine carcinoma of the breast. PLoS ONE 2014, 9, e112024. [Google Scholar] [CrossRef] [PubMed]
- Camarero, N.; Mascaro, C.; Mayordomo, C.; Vilardell, F.; Haro, D.; Marrero, P.F. Ketogenic hmgcs2 is a c-myc target gene expressed in differentiated cells of human colonic epithelium and down-regulated in colon cancer. Mol. Cancer Res. 2006, 4, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.E.; He, H.L.; Shiue, Y.L.; Lee, S.W.; Lin, L.C.; Wu, T.F.; Chang, I.W.; Lee, H.H.; Li, C.F. The prognostic impact of lipid biosynthesis-associated markers, hsd17b2 and hmgcs2, in rectal cancer treated with neoadjuvant concurrent chemoradiotherapy. Tumor Biol. 2015, 36, 7675–7683. [Google Scholar] [CrossRef] [PubMed]
- Prip-Buus, C.; Bouthillier-Voisin, A.C.; Kohl, C.; Demaugre, F.; Girard, J.; Pegorier, J.P. Evidence for an impaired long-chain fatty acid oxidation and ketogenesis in fao hepatoma cells. Eur. J. Biochem. 1992, 209, 291–298. [Google Scholar] [CrossRef]
- Su, S.G.; Yang, M.; Zhang, M.F.; Peng, Q.Z.; Li, M.Y.; Liu, L.P.; Bao, S.Y. Mir-107-mediated decrease of hmgcs2 indicates poor outcomes and promotes cell migration in hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 2017, 91, 53–59. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Lu, J.; Xia, Y.; Chen, K.; Zheng, Y.; Wang, J.; Lu, W.; Yin, Q.; Wang, F.; Zhou, Y.; Guo, C. Oncogenic role of the notch pathway in primary liver cancer. Oncol. Lett. 2016, 12, 3–10. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, P.A.; Carson, J.J.; Hebert, A.S.; Hubler, S.L.; Niemi, N.M.; Bailey, D.J.; Jochem, A.; Stapleton, D.S.; Keller, M.P.; Westphall, M.S.; et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012, 16, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Thumelin, S.; Forestier, M.; Girard, J.; Pegorier, J.P. Developmental changes in mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase gene expression in rat liver, intestine and kidney. Biochem. J. 1993, 292, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Le May, C.; Pineau, T.; Bigot, K.; Kohl, C.; Girard, J.; Pegorier, J.P. Reduced hepatic fatty acid oxidation in fasting pparalpha null mice is due to impaired mitochondrial hydroxymethylglutaryl-coa synthase gene expression. FEBS Lett. 2000, 475, 163–166. [Google Scholar] [CrossRef]
- Williamson, D.H.; Bates, M.W.; Krebs, H.A. Activity and intracellular distribution of enzymes of ketone-body metabolism in rat liver. Biochem. J. 1968, 108, 353–361. [Google Scholar] [CrossRef]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr.; Herrera, M.G.; Morgan, A.P.; Soeldner, J.S.; Steinke, J.; Levy, P.L.; Reichard, G.A., Jr.; Kipnis, D.M. Hormone-fuel interrelationships during fasting. J. Clin. Investig. 1966, 45, 1751–1769. [Google Scholar] [CrossRef]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Chriett, S.; Dabek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over beta-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Asher, J.L.; Molony, R.D.; Shaw, A.C.; Zeiss, C.J.; Wang, C.; Morozova-Roche, L.A.; Herzog, R.I.; Iwasaki, A.; Dixit, V.D. Beta-hydroxybutyrate deactivates neutrophil nlrp3 inflammasome to relieve gout flares. Cell Rep. 2017, 18, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Magee, B.A.; Potezny, N.; Rofe, A.M.; Conyers, R.A. The inhibition of malignant cell growth by ketone bodies. Aust. J. Exp. Biol. Med. Sci. 1979, 57, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Kostiuk, M.A.; Keller, B.O.; Berthiaume, L.G. Palmitoylation of ketogenic enzyme hmgcs2 enhances its interaction with pparalpha and transcription at the hmgcs2 ppre. FASEB J. 2010, 24, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.M.; Miyata, K.S.; Cechetto, J.D.; Rachubinski, R.A.; Capone, J.P. A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor pparalpha. EMBO J. 1998, 17, 6972–6978. [Google Scholar] [CrossRef] [PubMed]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: Challenges and opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar] [CrossRef]
- Xiao, Y.B.; Cai, S.H.; Liu, L.L.; Yang, X.; Yun, J.P. Decreased expression of peroxisome proliferator-activated receptor alpha indicates unfavorable outcomes in hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 1781–1789. [Google Scholar] [CrossRef]
- Tang, H.; Wu, Y.; Qin, Y.; Wang, H.; Jia, Y.; Yang, S.; Luo, S.; Wang, Q. Predictive significance of hmgcs2 for prognosis in resected chinese esophageal squamous cell carcinoma patients. Onco Targets Ther. 2017, 10, 2553–2560. [Google Scholar] [CrossRef][Green Version]
- Chen, S.W.; Chou, C.T.; Chang, C.C.; Li, Y.J.; Chen, S.T.; Lin, I.C.; Kok, S.H.; Cheng, S.J.; Lee, J.J.; Wu, T.S.; et al. Hmgcs2 enhances invasion and metastasis via direct interaction with pparalpha to activate src signaling in colorectal cancer and oral cancer. Oncotarget 2017, 8, 22460–22476. [Google Scholar]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef]
- Escrich, E.; Moral, R.; Grau, L.; Costa, I.; Solanas, M. Molecular mechanisms of the effects of olive oil and other dietary lipids on cancer. Mol. Nutr. Food Res. 2007, 51, 1279–1292. [Google Scholar] [CrossRef]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer 2008, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Mavropoulos, J.C.; Buschemeyer, W.C., 3rd; Tewari, A.K.; Rokhfeld, D.; Pollak, M.; Zhao, Y.; Febbo, P.G.; Cohen, P.; Hwang, D.; Devi, G.; et al. The effects of varying dietary carbohydrate and fat content on survival in a murine lncap prostate cancer xenograft model. Cancer Prev. Res. 2009, 2, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Healy, M.E.; Lahiri, S.; Hargett, S.R.; Chow, J.D.; Byrne, F.L.; Breen, D.S.; Kenwood, B.M.; Taddeo, E.P.; Lackner, C.; Caldwell, S.H.; et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci. Rep. 2016, 6, 22292. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Fang, C.C.; Yen, C.H.; Hsu, S.M.; Wang, C.K.; Huang, S.F.; Liang, Y.C.; Lin, Y.Y.; Chu, Y.T.; Chen, Y.M.A. Niemann-pick type c2 protein regulates liver cancer progression via modulating erk1/2 pathway: Clinicopathological correlations and therapeutical implications. Int. J. Cancer 2015, 137, 1341–1351. [Google Scholar] [CrossRef]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23306–23322. [Google Scholar] [CrossRef]
- Heindryckx, F.; Colle, I.; Van Vlierberghe, H. Experimental mouse models for hepatocellular carcinoma research. Int. J. Exp. Pathol. 2009, 90, 367–386. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-H.; Liu, C.-L.; Chiu, W.-C.; Twu, Y.-C.; Liao, Y.-J. HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma. Cancers 2019, 11, 1876. https://doi.org/10.3390/cancers11121876
Wang Y-H, Liu C-L, Chiu W-C, Twu Y-C, Liao Y-J. HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma. Cancers. 2019; 11(12):1876. https://doi.org/10.3390/cancers11121876
Chicago/Turabian StyleWang, Yuan-Hsi, Chao-Lien Liu, Wan-Chun Chiu, Yuh-Ching Twu, and Yi-Jen Liao. 2019. "HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma" Cancers 11, no. 12: 1876. https://doi.org/10.3390/cancers11121876
APA StyleWang, Y.-H., Liu, C.-L., Chiu, W.-C., Twu, Y.-C., & Liao, Y.-J. (2019). HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma. Cancers, 11(12), 1876. https://doi.org/10.3390/cancers11121876