Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma

 ,
,
Abstract
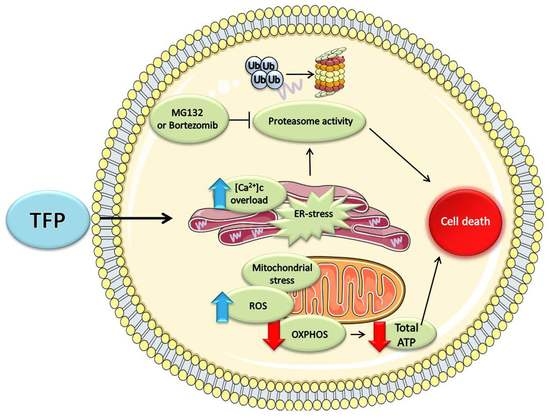
1. Introduction
2. Results
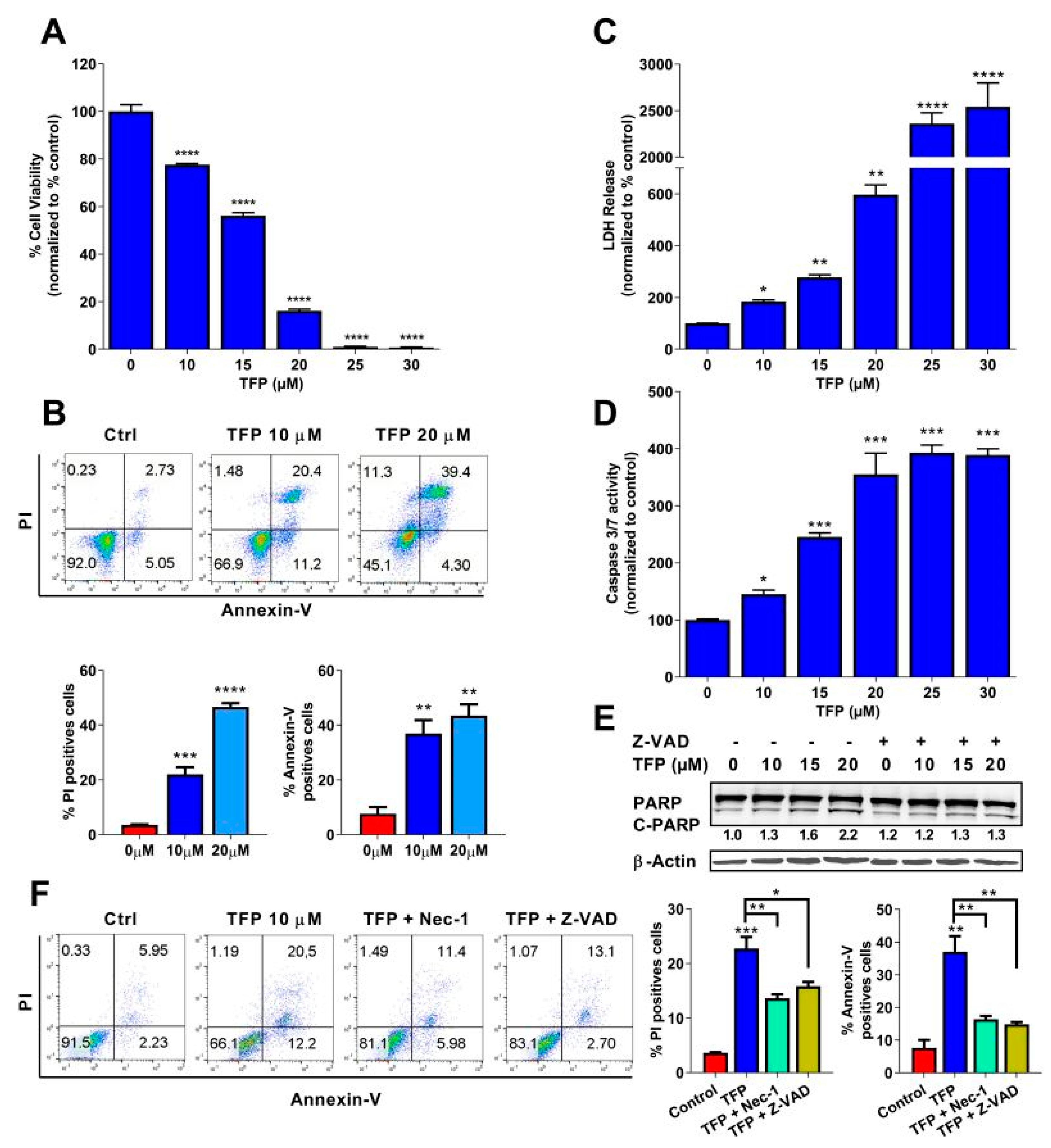
2.1. Trifluoperazine Induces Both Necrosis and Apoptosis in Pancreatic Cancer Cells
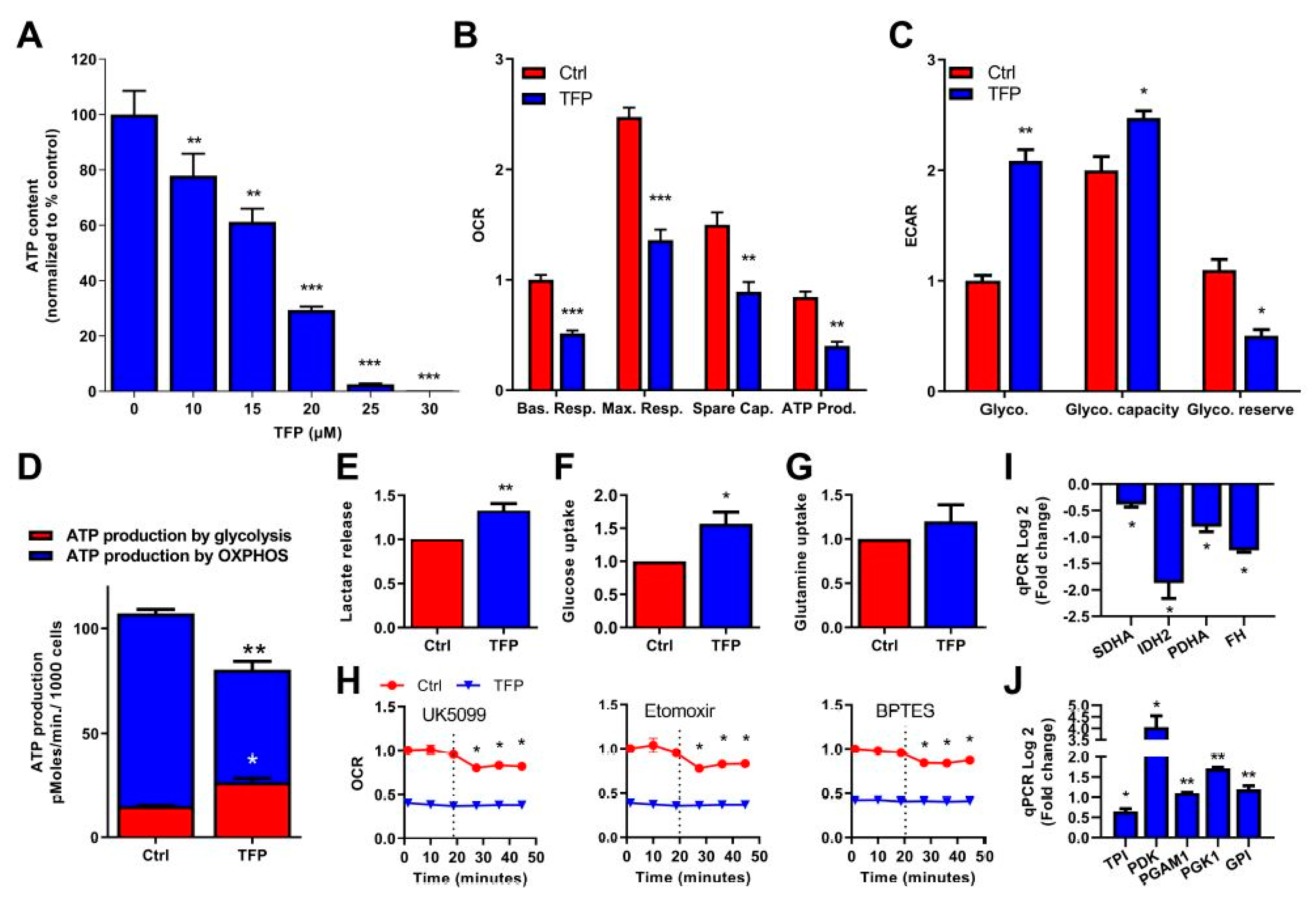
2.2. Trifluoperazine Decreases the Intracellular ATP Production
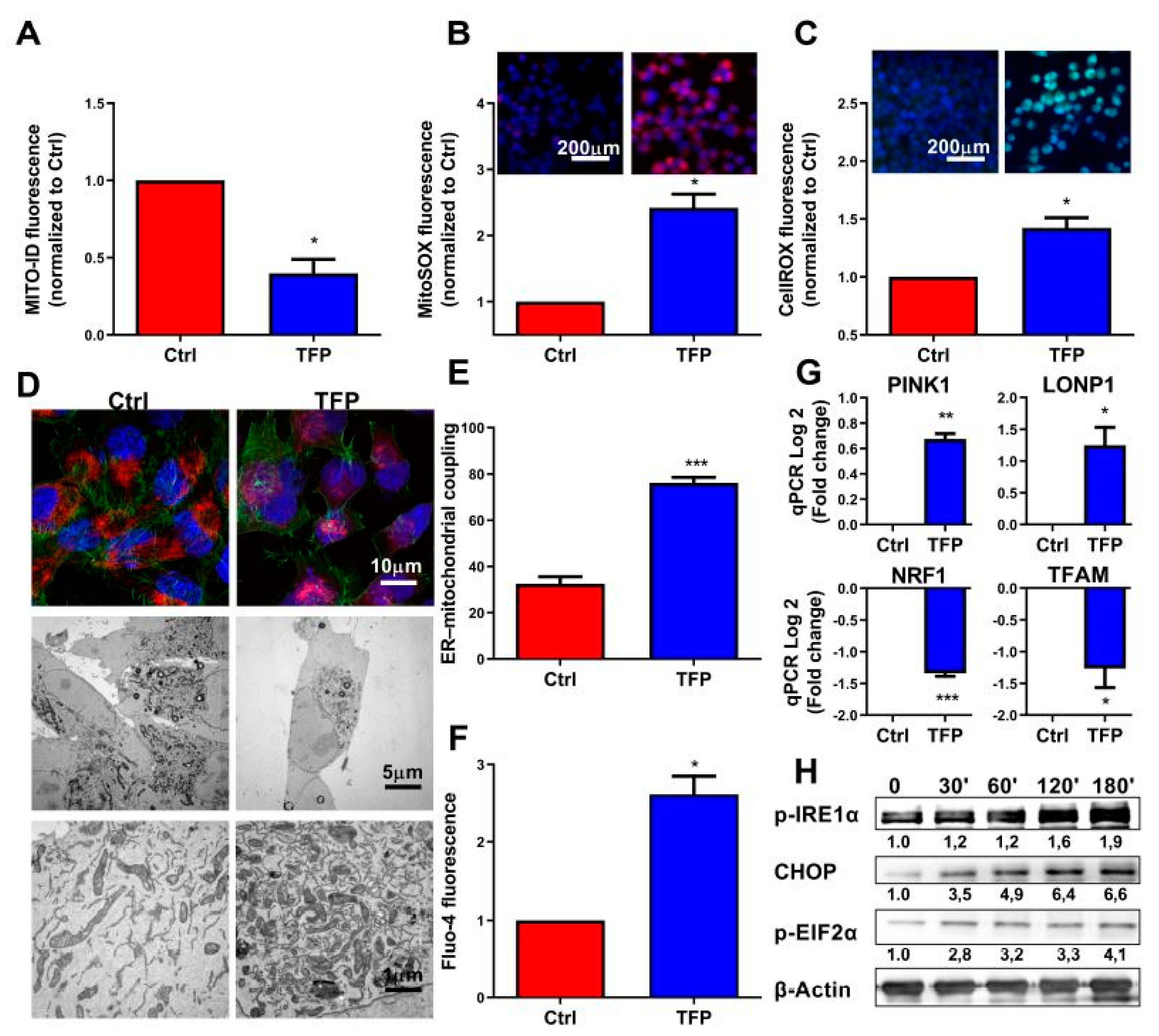
2.3. Trifluoperazine-treatment Induces Mitochondrial and Endoplasmic Coupled Stress
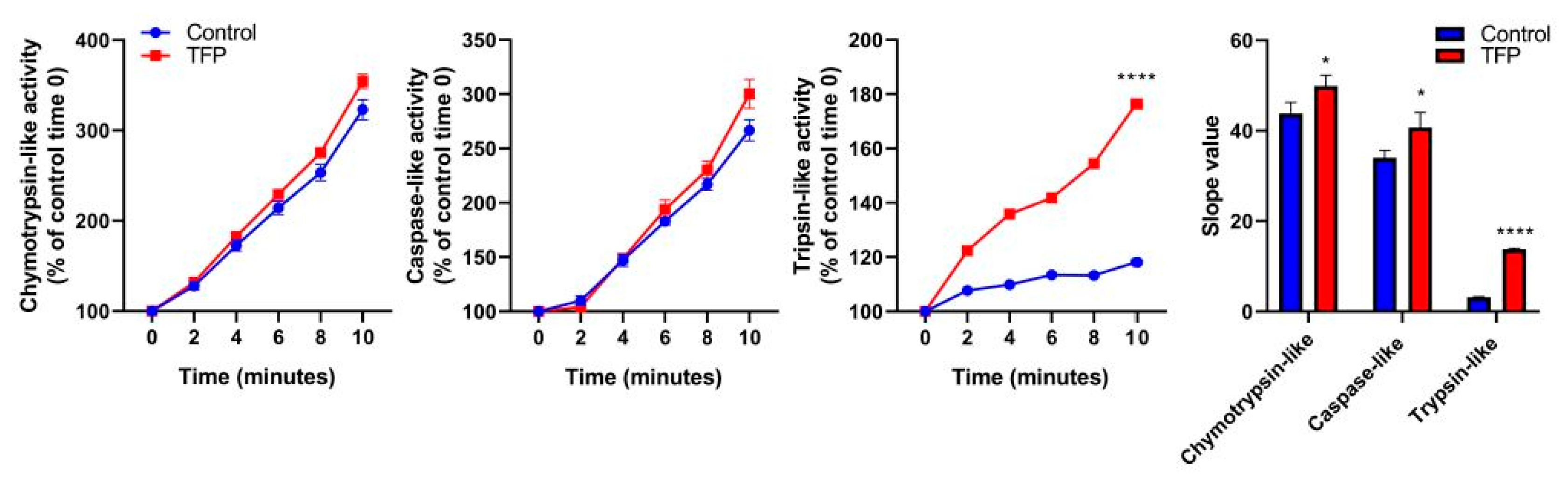
2.4. Proteasome System Activity was Activated after Trifluoperazine-treatment
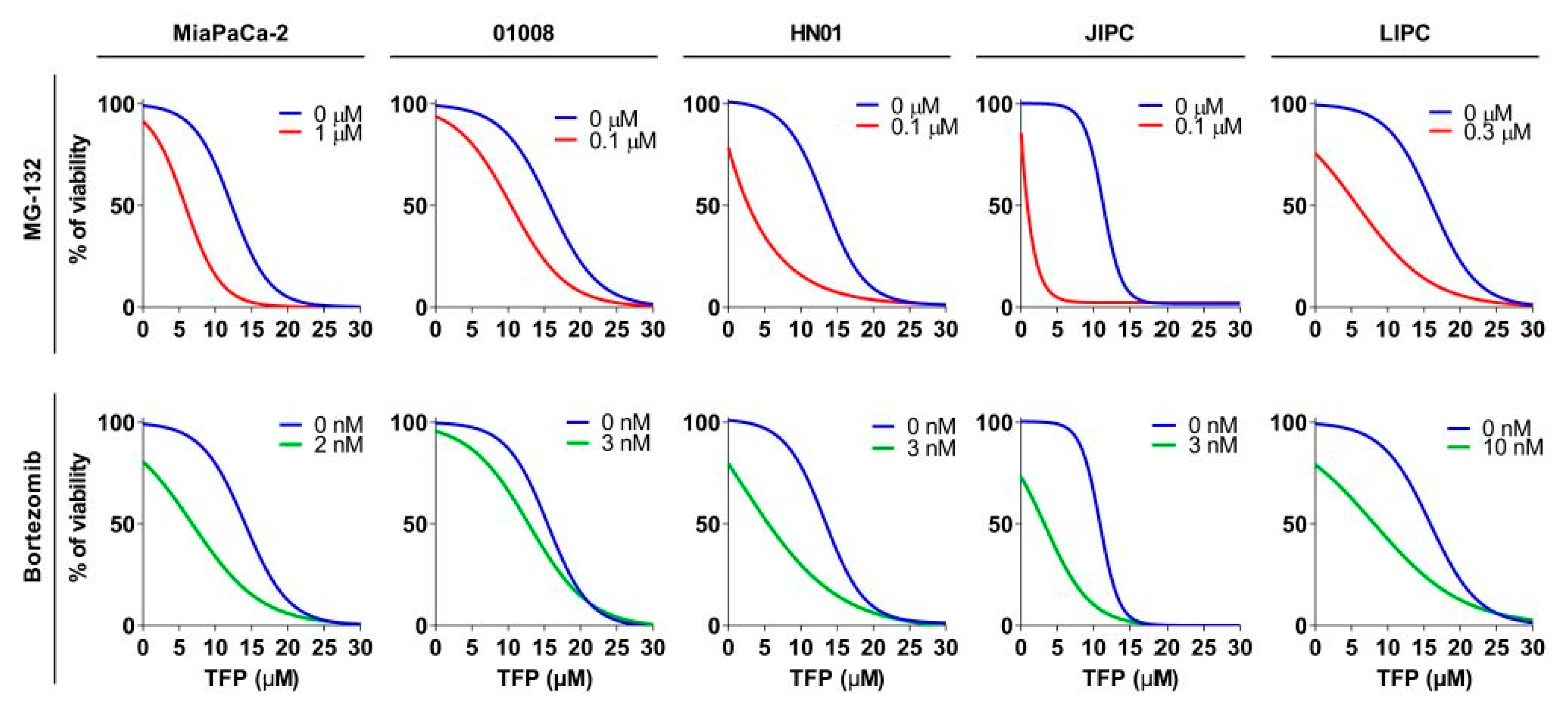
2.5. Proteasome Inhibition Increases Sensitivity to Trifluoperazine of Primary Pancreatic Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. LDH Assay, ATP Production, and Caspase-3/7 Activity Assay
4.3. qRT-PCR
4.4. Western Blot Analysis
4.5. Extracellular Flux Assay
4.6. Lactate Release, Glucose and Glutamine Uptake
4.7. Annexin V/PI Staining
4.8. ROS Measurements
4.9. Mitochondrial Membrane Potential
4.10. Cytosolic Calcium Concentration
4.11. Mitochondrial Network
4.12. Electron Microscopy
4.13. Chemograms and Viability Assays
4.14. Proteasome Activity Assay
4.15. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brunner, M.; Wu, Z.; Krautz, C.; Pilarsky, C.; Grützmann, R.; Weber, G.F. Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions. Int. J. Mol. Sci. 2019, 20, 4543. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Scapozza, L.; Altaba, A.R.I. Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 434–454. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a Well-Known Antipsychotic, Inhibits Glioblastoma Invasion by Binding to Calmodulin and Disinhibiting Calcium Release Channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Sun, L.; Zhou, G.; Ge, H.; Meng, Y.; Li, J.; Li, X.; Fang, X. Trifluoperazine as an alternative strategy for the inhibition of tumor growth of colorectal cancer. J. Cell Biochem. 2019, 120, 15756–15765. [Google Scholar] [CrossRef]
- Feng, Z.; Xia, Y.; Gao, T.; Xu, F.; Lei, Q.; Peng, C.; Yang, Y.; Xue, Q.; Hu, X.; Wang, Q.; et al. The antipsychotic agent trifluoperazine hydrochloride suppresses triple-negative breast cancer tumor growth and brain metastasis by inducing G0/G1 arrest and apoptosis. Cell Death Dis. 2018, 9, 1006. [Google Scholar] [CrossRef]
- Pulkoski-Gross, A.; Li, J.; Gao, T.; Xu, F.; Lei, Q.; Peng, C.; Yang, Y.; Xue, Q.; Hu, X.; Wang, Q.; et al. Repurposing the antipsychotic trifluoperazine as an antimetastasis agent. Mol. Pharmacol. 2015, 87, 501–512. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, Z.; Chen, X.; Luo, R.; Cai, H.; Wang, H.; Zhang, H.; Sun, T.; Zhang, Y. Trifluoperazine Activates FOXO1-Related Signals to Inhibit Tumor Growth in Hepatocellular Carcinoma. DNA Cell Biol. 2017, 36, 813–821. [Google Scholar] [CrossRef]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef]
- Yeh, C.T.; Wu, A.T.; Chang, P.M.; Chen, K.Y.; Yang, C.N.; Yang, S.C.; Ho, C.C.; Chen, C.C.; Kuo, Y.L.; Lee, P.Y.; et al. Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am. J. Respir. Crit. Care Med. 2012, 186, 1180–1188. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, R.; Zhang, C.; Xu, Y.; Han, M.; Huang, B.; Chen, A.; Qiu, C.; Thorsen, F.; Prestegarden, L.; et al. Trifluoperazine, a novel autophagy inhibitor, increases radiosensitivity in glioblastoma by impairing homologous recombination. J. Exp. Clin. Cancer Res. 2017, 36, 118. [Google Scholar] [CrossRef]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]
- Kato, H.; Nishitoh, H. Stress responses from the endoplasmic reticulum in cancer. Front. Oncol. 2015, 5, 93. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Kuo, K.L.; Liu, S.H.; Lin, W.C.; Hsu, F.S.; Chow, P.M.; Chang, Y.W.; Yang, S.P.; Shi, C.S.; Hsu, C.H.; Liao, S.M.; et al. Trifluoperazine, an Antipsychotic Drug, Effectively Reduces Drug Resistance in Cisplatin-Resistant Urothelial Carcinoma Cells via Suppressing Bcl-xL: An In Vitro and In Vivo Study. Int. J. Mol. Sci. 2019, 20, 3218. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Yang, W.L.; Lin, K.T.; Liu, C.H.; Liu, Y.W.; Huang, K.W.; Chang, P.M.; Lai, J.M.; Hsu, C.N.; Chao, K.M.; et al. Gene expression-based chemical genomics identifies potential therapeutic drugs in hepatocellular carcinoma. PLoS ONE 2011, 6, e27186. [Google Scholar] [CrossRef]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.M.; Johnson, J.; Mollaee, M.; Tassone, P.; Amin, D.; Knops, A.; Whitaker-Menezes, D.; Mahoney, M.G.; South, A.; Rodeck, U.; et al. Metformin Clinical Trial in HPV+ and HPV- Head and Neck Squamous Cell Carcinoma: Impact on Cancer Cell Apoptosis and Immune Infiltrate. Front. Oncol. 2018, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef]
- Shi, J.L.; Fu, L.; Wang, W.D. High expression of inositol 1, 4, 5-trisphosphate receptor, type 2 (ITPR2) as a novel biomarker for worse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget 2015, 6, 5299–5309. [Google Scholar] [CrossRef]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Chun, Y.S.; Lee, D.S.; Huang, L.E.; Park, J.W. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008, 111, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- Duconseil, P.; Gilabert, M.; Gayet, O.; Loncle, C.; Moutardier, V.; Turrini, O.; Calvo, E.; Ewald, J.; Giovannini, M.; Gasmi, M.; et al. Transcriptomic analysis predicts survival and sensitivity to anticancer drugs of patients with a pancreatic adenocarcinoma. Am. J. Pathol. 2015, 185, 1022–1032. [Google Scholar] [CrossRef]
- Wu, H.; Ying, M.; Hu, X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget 2016, 7, 40621–40629. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (µM) | AUC (a.u.) | ||||
|---|---|---|---|---|---|---|
| TFP | TFP + MG-132 | TFP + bortezomib | TFP | TFP + MG-132 | TFP + bortezomib | |
| MiaPaCa-2 | 13.24 ± 0.21 | 5.46 ± 0.17 **** | 6.81 ± 0.20 **** | 1372.67 ± 101.18 | 618.13 ± 49.26 **** | 939.37 ± 19.02 **** |
| 1008 | 10.46 ± 0.48 | 8.05 ± 0.10 ** | 8,65 ± 0.11 * | 1507.67 ± 49.97 | 1260.00 ± 10.78 ** | 1140.67 ± 15.16 ** |
| HN01 | 14.78 ± 0.044 | 2.47 ± 0.36 **** | 3.16 ± 0.29 **** | 1345.67 ± 15.026 | 525.7 ± 9.05 **** | 709.37 ± 24.60 **** |
| JIPC | 11.45 ± 0.067 | 0.86 ± 0,01 **** | 2.02 ± 0.44 **** | 1218 ± 14.98 | 242.5 ± 1.84 **** | 425.43 ± 34.75 **** |
| LIPC | 15.07 ± 8.07 | 5.81 ± 0.08 **** | 8.15 ± 0.09 **** | 1559 ± 28.583 | 754.1 ± 9.15 **** | 917 ± 16.49 **** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Lan, W.; Fraunhoffer, N.; Meilerman, A.; Iovanna, J.; Santofimia-Castaño, P. Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 1869. https://doi.org/10.3390/cancers11121869
Huang C, Lan W, Fraunhoffer N, Meilerman A, Iovanna J, Santofimia-Castaño P. Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma. Cancers. 2019; 11(12):1869. https://doi.org/10.3390/cancers11121869
Chicago/Turabian StyleHuang, Can, Wenjun Lan, Nicolas Fraunhoffer, Analía Meilerman, Juan Iovanna, and Patricia Santofimia-Castaño. 2019. "Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma" Cancers 11, no. 12: 1869. https://doi.org/10.3390/cancers11121869
APA StyleHuang, C., Lan, W., Fraunhoffer, N., Meilerman, A., Iovanna, J., & Santofimia-Castaño, P. (2019). Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma. Cancers, 11(12), 1869. https://doi.org/10.3390/cancers11121869