Elevated Autotaxin and LPA Levels during Chronic Viral Hepatitis and Hepatocellular Carcinoma Associate with Systemic Immune Activation

Abstract
1. Introduction
2. Autotaxin Is Elevated during Liver Disease, Including during Chronic HCV Infection
3. Elevated Plasma LPA Levels Are Present during HCV Infection and Likely HCC
4. Immune Activation and Morbidity in the Setting of Chronic Viral Infection and Aging
5. Elevated Levels of ATX during Chronic HCV Infection Are Associated with Systemic Immune Activation
6. Partial Normalization of ATX Levels within Months of Starting IFN-Free DAA HCV Therapy Is Associated with Variable Degrees of Normalization of Parameters of Systemic Immune Activation
7. Concluding Remarks
- ATX is elevated during a number of chronic liver disease states, most commonly associating with degree of liver fibrosis. Levels of ATX are most commonly associated with levels of the ATX enzymatic activity product, LPA, a lipid mediator that signals through LPA receptors.
- The ATX-LPA axis is associated with a number of biologic pathways including airway inflammation, wound healing, as well as a variety of cancers, and immune modulation. In regard to the latter, the ATX-LPA axis is associated with systemic immune activation in the settings of HCV and HCV/HIV infection.
- Since systemic immune activation is associated with morbidity and mortality during chronic viral infection, and since ATX-LPA signaling may contribute to immune activation, further investigation of the relationship between ATX-LPA signaling and morbidity during chronic viral infection is warranted.
- ATX plasma levels and markers of systemic immune activation partially normalize with HCV direct-acting antiviral therapy, indicating a non-fibrotic and reversible component contributing to elevated ATX levels during chronic hepatic viral infection.
Funding
Conflicts of Interest
References
- Stefan, C.; Jansen, S.; Bollen, M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem. Sci. 2005, 30, 542–550. [Google Scholar] [CrossRef]
- Giganti, A.; Rodriguez, M.; Fould, B.; Moulharat, N.; Coge, F.; Chomarat, P.; Galizzi, J.P.; Valet, P.; Saulnier-Blache, J.S.; Boutin, J.A.; et al. Murine and human autotaxin alpha, beta, and gamma isoforms: Gene organization, tissue distribution, and biochemical characterization. J. Biol. Chem. 2008, 283, 7776–7789. [Google Scholar] [CrossRef]
- Kanda, H.; Newton, R.; Klein, R.; Morita, Y.; Gunn, M.D.; Rosen, S.D. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 2008, 9, 415–423. [Google Scholar] [CrossRef]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Bai, Z.; Cai, L.; Umemoto, E.; Takeda, A.; Tohya, K.; Komai, Y.; Veeraveedu, P.T.; Hata, E.; Sugiura, Y.; Kubo, A.; et al. Constitutive lymphocyte transmigration across the basal lamina of high endothelial venules is regulated by the autotaxin/lysophosphatidic acid axis. J. Immunol. 2013, 190, 2036–2048. [Google Scholar] [CrossRef]
- Benesch, M.G.; Ko, Y.M.; Tang, X.; Dewald, J.; Lopez-Campistrous, A.; Zhao, Y.Y.; Lai, R.; Curtis, J.M.; Brindley, D.N.; McMullen, T.P. Autotaxin is an inflammatory mediator and therapeutic target in thyroid cancer. Endocr. Relat. Cancer 2015, 22, 593–607. [Google Scholar] [CrossRef]
- Cotte, A.K.; Cottet, V.; Aires, V.; Mouillot, T.; Rizk, M.; Vinault, S.; Binquet, C.; de Barros, J.P.; Hillon, P.; Delmas, D. Phospholipid profiles and hepatocellular carcinoma risk and prognosis in cirrhotic patients. Oncotarget 2019, 10, 2161–2172. [Google Scholar] [CrossRef]
- Fukushima, K.; Takahashi, K.; Yamasaki, E.; Onishi, Y.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Lysophosphatidic acid signaling via LPA1 and LPA3 regulates cellular functions during tumor progression in pancreatic cancer cells. Exp. Cell Res. 2017, 352, 139–145. [Google Scholar] [CrossRef]
- Kishi, Y.; Okudaira, S.; Tanaka, M.; Hama, K.; Shida, D.; Kitayama, J.; Yamori, T.; Aoki, J.; Fujimaki, T.; Arai, H. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J. Biol. Chem. 2006, 281, 17492–17500. [Google Scholar] [CrossRef]
- Li, Y.Y.; Zhang, W.C.; Zhang, J.L.; Zheng, C.J.; Zhu, H.; Yu, H.M.; Fan, L.M. Plasma levels of lysophosphatidic acid in ovarian cancer versus controls: A meta-analysis. Lipids Health Dis. 2015, 14, 72. [Google Scholar] [CrossRef]
- Liao, Y.; Mu, G.; Zhang, L.; Zhou, W.; Zhang, J.; Yu, H. Lysophosphatidic acid stimulates activation of focal adhesion kinase and paxillin and promotes cell motility, via LPA1-3, in human pancreatic cancer. Dig. Dis. Sci. 2013, 58, 3524–3533. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef]
- Zhao, Y.; Natarajan, V. Lysophosphatidic acid signaling in airway epithelium: Role in airway inflammation and remodeling. Cell. Signal. 2009, 21, 367–377. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef]
- Aidinis, V.; Carninci, P.; Armaka, M.; Witke, W.; Harokopos, V.; Pavelka, N.; Koczan, D.; Argyropoulos, C.; Thwin, M.M.; Moller, S.; et al. Cytoskeletal rearrangements in synovial fibroblasts as a novel pathophysiological determinant of modeled rheumatoid arthritis. PLoS Genet. 2005, 1, e48. [Google Scholar] [CrossRef]
- Ikeda, H.; Yatomi, Y.; Yanase, M.; Satoh, H.; Nishihara, A.; Kawabata, M.; Fujiwara, K. Effects of lysophosphatidic acid on proliferation of stellate cells and hepatocytes in culture. Biochem. Biophys. Res. Commun. 1998, 248, 436–440. [Google Scholar] [CrossRef]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Yanase, M.; Ikeda, H.; Matsui, A.; Maekawa, H.; Noiri, E.; Tomiya, T.; Arai, M.; Yano, T.; Shibata, M.; Ikebe, M.; et al. Lysophosphatidic acid enhances collagen gel contraction by hepatic stellate cells: Association with rho-kinase. Biochem. Biophys. Res. Commun. 2000, 277, 72–78. [Google Scholar] [CrossRef]
- Fujimori, N.; Umemura, T.; Kimura, T.; Tanaka, N.; Sugiura, A.; Yamazaki, T.; Joshita, S.; Komatsu, M.; Usami, Y.; Sano, K.; et al. Serum autotaxin levels are correlated with hepatic fibrosis and ballooning in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2018, 24, 1239–1249. [Google Scholar] [CrossRef]
- Kostadinova, L.; Shive, C.; Judge, C.; Zebrowski, E.; Compan, A.; Rife, K.; Hirsch, A.; Falck-Ytter, Y.; Schlatzer, D.; Li, X.; et al. During HCV and HCV-HIV infection elevated plasma Autotaxin is associated with LPA and markers of immune activation that normalize during IFN-free HCV therapy. J. Infect. Dis. 2016. [Google Scholar] [CrossRef]
- Wunsch, E.; Krawczyk, M.; Milkiewicz, M.; Trottier, J.; Barbier, O.; Neurath, M.F.; Lammert, F.; Kremer, A.E.; Milkiewicz, P. Serum Autotaxin is a Marker of the Severity of Liver Injury and Overall Survival in Patients with Cholestatic Liver Diseases. Sci. Rep. 2016, 6, 30847. [Google Scholar] [CrossRef]
- Joshita, S.; Ichikawa, Y.; Umemura, T.; Usami, Y.; Sugiura, A.; Shibata, S.; Yamazaki, T.; Fujimori, N.; Komatsu, M.; Matsumoto, A.; et al. Serum autotaxin is a useful liver fibrosis marker in patients with chronic hepatitis B virus infection. Hepatol. Res. 2018, 48, 275–285. [Google Scholar] [CrossRef]
- Kostadinova, L.; Shive, C.L.; Zebrowski, E.; Fuller, B.; Rife, K.; Hirsch, A.; Compan, A.; Moreland, A.; Falck-Ytter, Y.; Popkin, D.L.; et al. Soluble Markers of Immune Activation Differentially Normalize and Selectively Associate with Improvement in AST, ALT, Albumin, and Transient Elastography during IFN-Free HCV Therapy. Pathog. Immun. 2018, 3, 149–163. [Google Scholar] [CrossRef]
- Pleli, T.; Martin, D.; Kronenberger, B.; Brunner, F.; Koberle, V.; Grammatikos, G.; Farnik, H.; Martinez, Y.; Finkelmeier, F.; Labocha, S.; et al. Serum autotaxin is a parameter for the severity of liver cirrhosis and overall survival in patients with liver cirrhosis--a prospective cohort study. PLoS ONE 2014, 9, e103532. [Google Scholar] [CrossRef]
- Rachakonda, V.P.; Reeves, V.L.; Aljammal, J.; Wills, R.C.; Trybula, J.S.; DeLany, J.P.; Kienesberger, P.C.; Kershaw, E.E. Serum autotaxin is independently associated with hepatic steatosis in women with severe obesity. Obesity (Silver Spring) 2015, 23, 965–972. [Google Scholar] [CrossRef]
- Yamazaki, T.; Joshita, S.; Umemura, T.; Usami, Y.; Sugiura, A.; Fujimori, N.; Shibata, S.; Ichikawa, Y.; Komatsu, M.; Matsumoto, A.; et al. Association of Serum Autotaxin Levels with Liver Fibrosis in Patients with Chronic Hepatitis, C. Sci. Rep. 2017, 7, 46705. [Google Scholar] [CrossRef]
- Dhillon, A.K.; Kremer, A.E.; Kummen, M.; Boberg, K.M.; Elferink, R.P.O.; Karlsen, T.H.; Beuers, U.; Vesterhus, M.; Hov, J.R. Autotaxin activity predicts transplant-free survival in primary sclerosing cholangitis. Sci. Rep. 2019, 9, 8450. [Google Scholar] [CrossRef]
- Honda, Y.; Imajo, K.; Kobayashi, T.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Yoneda, M.; Kobayashi, N.; Saito, S.; Nakajima, A. Autotaxin is a valuable biomarker for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. Hepatol. Res. 2019. [Google Scholar] [CrossRef]
- Kondo, M.; Ishizawa, T.; Enooku, K.; Tokuhara, Y.; Ohkawa, R.; Uranbileg, B.; Nakagawa, H.; Tateishi, R.; Yoshida, H.; Kokudo, N.; et al. Increased serum autotaxin levels in hepatocellular carcinoma patients were caused by background liver fibrosis but not by carcinoma. Clin. Chim. Acta 2014, 433, 128–134. [Google Scholar] [CrossRef]
- Enooku, K.; Uranbileg, B.; Ikeda, H.; Kurano, M.; Sato, M.; Kudo, H.; Maki, H.; Koike, K.; Hasegawa, K.; Kokudo, N.; et al. Higher LPA2 and LPA6 mRNA Levels in Hepatocellular Carcinoma Are Associated with Poorer Differentiation, Microvascular Invasion and Earlier Recurrence with Higher Serum Autotaxin Levels. PLoS ONE 2016, 11, e0161825. [Google Scholar] [CrossRef]
- Jansen, S.; Andries, M.; Vekemans, K.; Vanbilloen, H.; Verbruggen, A.; Bollen, M. Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells. Cancer Lett. 2009, 284, 216–221. [Google Scholar] [CrossRef]
- Muro, H.; Shirasawa, H.; Kosugi, I.; Nakamura, S. Defect of Fc receptors and phenotypical changes in sinusoidal endothelial cells in human liver cirrhosis. Am. J. Pathol. 1993, 143, 105–120. [Google Scholar]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef]
- Wu, J.M.; Xu, Y.; Skill, N.J.; Sheng, H.; Zhao, Z.; Yu, M.; Saxena, R.; Maluccio, M.A. Autotaxin expression and its connection with the TNF-alpha-NF-kappaB axis in human hepatocellular carcinoma. Mol. Cancer 2010, 9, 71. [Google Scholar] [CrossRef]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid. Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef]
- Wang, D.; Liem, D.A.; Lau, E.; Ng, D.C.; Bleakley, B.J.; Cadeiras, M.; Deng, M.C.; Lam, M.P.; Ping, P. Characterization of human plasma proteome dynamics using deuterium oxide. Proteom. Clin. Appl. 2014, 8, 610–619. [Google Scholar] [CrossRef]
- Nakamura, K.; Igarashi, K.; Ide, K.; Ohkawa, R.; Okubo, S.; Yokota, H.; Masuda, A.; Oshima, N.; Takeuchi, T.; Nangaku, M.; et al. Validation of an autotaxin enzyme immunoassay in human serum samples and its application to hypoalbuminemia differentiation. Clin. Chim. Acta 2008, 388, 51–58. [Google Scholar] [CrossRef]
- Nakamura, K.; Ohkawa, R.; Okubo, S.; Tozuka, M.; Okada, M.; Aoki, S.; Aoki, J.; Arai, H.; Ikeda, H.; Yatomi, Y. Measurement of lysophospholipase D/autotaxin activity in human serum samples. Clin. Biochem. 2007, 40, 274–277. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Cooper, A.B.; Wu, J.; Lu, D.; Maluccio, M.A. Is autotaxin (ENPP2) the link between hepatitis C and hepatocellular cancer? J. Gastrointest. Surg. 2007, 11, 1628–1634, discussion 1634-1625. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, K.J.; Panupinthu, N.; Yu, S.; Lee, J.; Han, J.W.; Kim, J.M.; Lee, J.S.; Kang, J.; Park, C.G.; et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011, 30, 1351–1359. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Z.; Xu, S.; Ni, L.; Wang, X. Expression of autotaxin mRNA in human hepatocellular carcinoma. Chin. Med. J. (Engl.) 1999, 112, 330–332. [Google Scholar]
- Watanabe, N.; Ikeda, H.; Nakamura, K.; Ohkawa, R.; Kume, Y.; Aoki, J.; Hama, K.; Okudaira, S.; Tanaka, M.; Tomiya, T.; et al. Both plasma lysophosphatidic acid and serum autotaxin levels are increased in chronic hepatitis C. J. Clin. Gastroenterol. 2007, 41, 616–623. [Google Scholar] [CrossRef]
- Mazzocca, A.; Dituri, F.; Lupo, L.; Quaranta, M.; Antonaci, S.; Giannelli, G. Tumor-secreted lysophostatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology 2011, 54, 920–930. [Google Scholar] [CrossRef]
- Skill, N.J.; Jianmin, W.; Yan, X.; Zhao, Z.; Tector, A.J.; Maluccio, M.A. Lysophospholipid variants in hepatocellular carcinoma. J. Surg. Res. 2013, 182, 241–249. [Google Scholar] [CrossRef]
- Kihara, Y.; Mizuno, H.; Chun, J. Lysophospholipid receptors in drug discovery. Exp. Cell Res. 2015, 333, 171–177. [Google Scholar] [CrossRef]
- Sokolov, E.; Eheim, A.L.; Ahrens, W.A.; Walling, T.L.; Swet, J.H.; McMillan, M.T.; Simo, K.A.; Thompson, K.J.; Sindram, D.; McKillop, I.H. Lysophosphatidic acid receptor expression and function in human hepatocellular carcinoma. J. Surg. Res. 2013, 180, 104–113. [Google Scholar] [CrossRef]
- Furman, D.; Chang, J.; Lartigue, L.; Bolen, C.R.; Haddad, F.; Gaudilliere, B.; Ganio, E.A.; Fragiadakis, G.K.; Spitzer, M.H.; Douchet, I.; et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 2017, 23, 174–184. [Google Scholar] [CrossRef]
- Hunt, P.W.; Sinclair, E.; Rodriguez, B.; Shive, C.; Clagett, B.; Funderburg, N.; Robinson, J.; Huang, Y.; Epling, L.; Martin, J.N.; et al. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J. Infect. Dis. 2014, 210, 1228–1238. [Google Scholar] [CrossRef]
- Tenorio, A.R.; Zheng, Y.; Bosch, R.J.; Krishnan, S.; Rodriguez, B.; Hunt, P.W.; Plants, J.; Seth, A.; Wilson, C.C.; Deeks, S.G.; et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J. Infect. Dis. 2014, 210, 1248–1259. [Google Scholar] [CrossRef]
- Wikby, A.; Nilsson, B.O.; Forsey, R.; Thompson, J.; Strindhall, J.; Lofgren, S.; Ernerudh, J.; Pawelec, G.; Ferguson, F.; Johansson, B. The immune risk phenotype is associated with IL-6 in the terminal decline stage: Findings from the Swedish NONA immune longitudinal study of very late life functioning. Mech. Ageing Dev. 2006, 127, 695–704. [Google Scholar] [CrossRef]
- Zampino, R.; Marrone, A.; Restivo, L.; Guerrera, B.; Sellitto, A.; Rinaldi, L.; Romano, C.; Adinolfi, L.E. Chronic HCV infection and inflammation: Clinical impact on hepatic and extra-hepatic manifestations. World J. Hepatol. 2013, 5, 528–540. [Google Scholar] [CrossRef]
- Ershler, W.B.; Keller, E.T. Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu. Rev. Med. 2000, 51, 245–270. [Google Scholar] [CrossRef]
- Harris, T.B.; Ferrucci, L.; Tracy, R.P.; Corti, M.C.; Wacholder, S.; Ettinger, W.H., Jr.; Heimovitz, H.; Cohen, H.J.; Wallace, R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am. J. Med. 1999, 106, 506–512. [Google Scholar]
- Sandler, N.G.; Koh, C.; Roque, A.; Eccleston, J.L.; Siegel, R.B.; Demino, M.; Kleiner, D.E.; Deeks, S.G.; Liang, T.J.; Heller, T.; et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 2011, 141, 1220–1230. [Google Scholar] [CrossRef]
- Pan, Z.; Zhou, L.; Hetherington, C.J.; Zhang, D.E. Hepatocytes contribute to soluble CD14 production, and CD14 expression is differentially regulated in hepatocytes and monocytes. J. Biol. Chem. 2000, 275, 36430–36435. [Google Scholar] [CrossRef]
- Shive, C.L.; Jiang, W.; Anthony, D.D.; Lederman, M.M. Soluble CD14 is a nonspecific marker of monocyte activation. AIDS 2015, 29, 1263–1265. [Google Scholar] [CrossRef]
- Reiner, A.P.; Lange, E.M.; Jenny, N.S.; Chaves, P.H.; Ellis, J.; Li, J.; Walston, J.; Lange, L.A.; Cushman, M.; Tracy, R.P. Soluble CD14: Genomewide association analysis and relationship to cardiovascular risk and mortality in older adults. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 158–164. [Google Scholar] [CrossRef]
- Kazankov, K.; Barrera, F.; Moller, H.J.; Bibby, B.M.; Vilstrup, H.; George, J.; Gronbaek, H. Soluble CD163, a macrophage activation marker, is independently associated with fibrosis in patients with chronic viral hepatitis B and C. Hepatology 2014, 60, 521–530. [Google Scholar] [CrossRef]
- Kuniholm, M.H.; Hanna, D.B.; Landay, A.L.; Kaplan, R.C.; Ley, K. Soluble CD163 is associated with noninvasive measures of liver fibrosis in hepatitis C virus- and hepatitis C virus/human immunodeficiency virus-infected women. Hepatology 2015, 61, 734–735. [Google Scholar] [CrossRef]
- Lidofsky, A.; Holmes, J.A.; Feeney, E.R.; Kruger, A.J.; Salloum, S.; Zheng, H.; Seguin, I.S.; Altinbas, A.; Masia, R.; Corey, K.E.; et al. Macrophage Activation Marker Soluble CD163 Is a Dynamic Marker of Liver Fibrogenesis in Human Immunodeficiency Virus/Hepatitis C Virus Coinfection. J. Infect. Dis. 2018, 218, 1394–1403. [Google Scholar] [CrossRef]
- Shmagel, K.V.; Saidakova, E.V.; Shmagel, N.G.; Korolevskaya, L.B.; Chereshnev, V.A.; Robinson, J.; Grivel, J.C.; Douek, D.C.; Margolis, L.; Anthony, D.D.; et al. Systemic inflammation and liver damage in HIV/hepatitis C virus coinfection. HIV Med. 2016. [Google Scholar] [CrossRef]
- Liang, H.; Duan, Z.; Li, D.; Li, D.; Wang, Z.; Ren, L.; Shen, T.; Shao, Y. Higher levels of circulating monocyte-platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell. Mol. Immunol. 2015, 12, 435–443. [Google Scholar] [CrossRef]
- Allen, N.; Barrett, T.J.; Guo, Y.; Nardi, M.; Ramkhelawon, B.; Rockman, C.B.; Hochman, J.S.; Berger, J.S. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis 2019, 282, 11–18. [Google Scholar] [CrossRef]
- Burdo, T.H.; Lo, J.; Abbara, S.; Wei, J.; DeLelys, M.E.; Preffer, F.; Rosenberg, E.S.; Williams, K.C.; Grinspoon, S. Soluble CD163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in HIV-infected patients. J. Infect. Dis. 2011, 204, 1227–1236. [Google Scholar] [CrossRef]
- Mascia, C.; Lichtner, M.; Zuccala, P.; Vita, S.; Tieghi, T.; Marocco, R.; Savinelli, S.; Rossi, R.; Iannetta, M.; Campagna, M.; et al. Active HCV infection is associated with increased circulating levels of interferon-gamma (IFN-gamma)-inducible protein-10 (IP-10), soluble CD163 and inflammatory monocytes regardless of liver fibrosis and HIV coinfection. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 644–655. [Google Scholar] [CrossRef]
- Shive, C.L.; Judge, C.J.; Clagett, B.; Kalayjian, R.C.; Osborn, M.; Sherman, K.E.; Fichtenbaum, C.; Gandhi, R.T.; Kang, M.; Popkin, D.L.; et al. Pre-vaccine plasma levels of soluble inflammatory indices negatively predict responses to HAV, HBV, and tetanus vaccines in HCV and HIV infection. Vaccine 2018, 36, 453–460. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, T.; Wang, R.; Zhang, H.; Huang, X.; Yin, J.; Zhang, L.; Xu, X.; Wu, H. Plasma IP-10 is associated with rapid disease progression in early HIV-1 infection. Viral. Immunol. 2012, 25, 333–337. [Google Scholar] [CrossRef]
- Liovat, A.S.; Rey-Cuille, M.A.; Lecuroux, C.; Jacquelin, B.; Girault, I.; Petitjean, G.; Zitoun, Y.; Venet, A.; Barre-Sinoussi, F.; Lebon, P.; et al. Acute plasma biomarkers of T cell activation set-point levels and of disease progression in HIV-1 infection. PLoS ONE 2012, 7, e46143. [Google Scholar] [CrossRef]
- Janssen, S.P.; Gayan-Ramirez, G.; Van den Bergh, A.; Herijgers, P.; Maes, K.; Verbeken, E.; Decramer, M. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation 2005, 111, 996–1005. [Google Scholar] [CrossRef]
- Isoda, K.; Ohsuzu, F. The effect of interleukin-1 receptor antagonist on arteries and cholesterol metabolism. J. Atheroscler. Thromb. 2006, 13, 21–30. [Google Scholar] [CrossRef]
- McKibben, R.A.; Margolick, J.B.; Grinspoon, S.; Li, X.; Palella, F.J., Jr.; Kingsley, L.A.; Witt, M.D.; George, R.T.; Jacobson, L.P.; Budoff, M.; et al. Elevated levels of monocyte activation markers are associated with subclinical atherosclerosis in men with and those without HIV infection. J. Infect. Dis. 2015, 211, 1219–1228. [Google Scholar] [CrossRef]
- Yan, A.T.; Yan, R.T.; Cushman, M.; Redheuil, A.; Tracy, R.P.; Arnett, D.K.; Rosen, B.D.; McClelland, R.L.; Bluemke, D.A.; Lima, J.A. Relationship of interleukin-6 with regional and global left-ventricular function in asymptomatic individuals without clinical cardiovascular disease: Insights from the Multi-Ethnic Study of Atherosclerosis. Eur. Heart. J. 2010, 31, 875–882. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Buxton, J.A.; Kim, J.H. Hepatitis A and hepatitis B vaccination responses in persons with chronic hepatitis C infections: A review of the evidence and current recommendations. Can. J. Infect. Dis. Med. Microbiol. 2008, 19, 197–202. [Google Scholar]
- Cruciani, M.; Mengoli, C.; Serpelloni, G.; Lanza, A.; Gomma, M.; Nardi, S.; Rimondo, C.; Bricolo, F.; Consolaro, S.; Trevisan, M.; et al. Serologic response to hepatitis B vaccine with high dose and increasing number of injections in HIV infected adult patients. Vaccine 2009, 27, 17–22. [Google Scholar] [CrossRef]
- Haynes, L.; Swain, S.L. Why aging T cells fail: Implications for vaccination. Immunity 2006, 24, 663–666. [Google Scholar] [CrossRef]
- Merani, S.; Pawelec, G.; Kuchel, G.A.; McElhaney, J.E. Impact of Aging and Cytomegalovirus on Immunological Response to Influenza Vaccination and Infection. Front. Immunol. 2017, 8, 784. [Google Scholar] [CrossRef]
- Bartek, J.; Hodny, Z.; Lukas, J. Cytokine loops driving senescence. Nat. Biol. 2008, 10, 887–889. [Google Scholar] [CrossRef]
- Shive, C.L.; Clagett, B.; McCausland, M.R.; Mudd, J.C.; Funderburg, N.T.; Freeman, M.L.; Younes, S.; Ferrari, B.M.; Rodriguez, B.; McComsey, G.A.; et al. Inflammation perturbs the IL-7 axis, promoting senescence and exhaustion that broadly characterize immune failure in treated HIV infection. J. Acquir. Immune Defic. Syndr. 2015. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Song, Y.; Wang, B.; Song, R.; GHao, Y.; Wang, D.; GLi, Y.; GKong, Y. T-cell Immunoglobulin and ITIM Domain Contributes to CD8+ T-cell Immunosenescence. Aging Cell 2017, 17, e12716. [Google Scholar]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Zhang, Z.N.; Zhu, M.L.; Chen, Y.H.; Fu, Y.J.; Zhang, T.W.; Jiang, Y.J.; Chu, Z.X.; Shang, H. Elevation of Tim-3 and PD-1 expression on T cells appears early in HIV infection, and differential Tim-3 and PD-1 expression patterns can be induced by common gamma -chain cytokines. BioMed. Res. Int. 2015, 2015, 916936. [Google Scholar] [CrossRef]
- Moorman, J.P.; Zhang, C.L.; Ni, L.; Ma, C.J.; Zhang, Y.; Wu, X.Y.; Thayer, P.; Islam, T.M.; Borthwick, T.; Yao, Z.Q. Impaired hepatitis B vaccine responses during chronic hepatitis C infection: Involvement of the PD-1 pathway in regulating CD4(+) T cell responses. Vaccine 2011, 29, 3169–3176. [Google Scholar] [CrossRef]
- Shi, L.; Wang, J.M.; Ren, J.P.; Cheng, Y.Q.; Ying, R.S.; Wu, X.Y.; Lin, S.M.; Griffin, J.W.; Li, G.Y.; Moorman, J.P.; et al. KLRG1 impairs CD4+ T cell responses via p16ink4a and p27kip1 pathways: Role in hepatitis B vaccine failure in individuals with hepatitis C virus infection. J. Immunol. 2014, 192, 649–657. [Google Scholar] [CrossRef]
- Ansaldi, F.; Orsi, A.; Sticchi, L.; Bruzzone, B.; Icardi, G. Hepatitis C virus in the new era: Perspectives in epidemiology, prevention, diagnostics and predictors of response to therapy. World J. Gastroenterol. 2014, 20, 9633–9652. [Google Scholar] [CrossRef]
- Kaffe, E.; Magkrioti, C.; Aidinis, V. Deregulated Lysophosphatidic Acid Metabolism and Signaling in Liver Cancer. Cancers (Basel) 2019, 11, 1262. [Google Scholar] [CrossRef]
- Im, E.; Motiejunaite, R.; Aranda, J.; Park, E.Y.; Federico, L.; Kim, T.I.; Clair, T.; Stracke, M.L.; Smyth, S.; Kazlauskas, A. Phospholipase Cgamma activation drives increased production of autotaxin in endothelial cells and lysophosphatidic acid-dependent regression. Mol. Cell. Biol. 2010, 30, 2401–2410. [Google Scholar] [CrossRef]
- Bao, L.; Qi, J.; Wang, Y.W.; Xi, Q.; Tserennadmid, T.; Zhao, P.F.; Qi, J.; Damirin, A. The atherogenic actions of LPC on vascular smooth muscle cells and its LPA receptor mediated mechanism. Biochem. Biophys. Res. Commun. 2018, 503, 1911–1918. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J. Lipopolysaccharide induces autotaxin expression in human monocytic THP-1 cells. Biochem. Biophys. Res. Commun. 2009, 378, 264–268. [Google Scholar] [CrossRef]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef]
- Siess, W.; Zangl, K.J.; Essler, M.; Bauer, M.; Brandl, R.; Corrinth, C.; Bittman, R.; Tigyi, G.; Aepfelbacher, M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6931–6936. [Google Scholar] [CrossRef]
- Chang, C.L.; Hsu, H.Y.; Lin, H.Y.; Chiang, W.; Lee, H. Lysophosphatidic acid-induced oxidized low-density lipoprotein uptake is class A scavenger receptor-dependent in macrophages. Prostaglandins Lipid Mediat. 2008, 87, 20–25. [Google Scholar] [CrossRef]
- Fueller, M.; Wang, D.A.; Tigyi, G.; Siess, W. Activation of human monocytic cells by lysophosphatidic acid and sphingosine-1-phosphate. Cell. Signal. 2003, 15, 367–375. [Google Scholar] [CrossRef]
- D’Aquilio, F.; Procaccini, M.; Izzi, V.; Chiurchiu, V.; Giambra, V.; Carotenuto, F.; Di Nardo, P.; Baldini, P.M. Activatory properties of lysophosphatidic acid on human THP-1 cells. Inflammation 2007, 30, 167–177. [Google Scholar] [CrossRef]
- Chang, C.L.; Lin, M.E.; Hsu, H.Y.; Yao, C.L.; Hwang, S.M.; Pan, C.Y.; Hsu, C.Y.; Lee, H. Lysophosphatidic acid-induced interleukin-1 beta expression is mediated through Gi/Rho and the generation of reactive oxygen species in macrophages. J. Biomed. Sci. 2008, 15, 357–363. [Google Scholar] [CrossRef]
- Ray, R.; Rai, V. Lysophosphatidic acid converts monocytes into macrophages in both mice and humans. Blood 2017, 129, 1177–1183. [Google Scholar] [CrossRef]
- Zhou, Z.B.; Yang, B.; Li, X.; Liu, H.; Lei, G. Lysophosphatidic Acid Promotes Expression and Activation of Matrix Metalloproteinase 9 (MMP9) in THP-1 Cells via Toll-Like Receptor 4/Nuclear Factor-kappaB (TLR4/NF-kappaB) Signaling Pathway. Med. Sci. Monit. 2018, 24, 4861–4868. [Google Scholar] [CrossRef]
- Liang, T.J.; Rehermann, B.; Seeff, L.B.; Hoofnagle, J.H. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. 2000, 132, 296–305. [Google Scholar] [CrossRef]
- Bility, M.T.; Nio, K.; Li, F.; McGivern, D.R.; Lemon, S.M.; Feeney, E.R.; Chung, R.T.; Su, L. Chronic hepatitis C infection-induced liver fibrogenesis is associated with M2 macrophage activation. Sci. Rep. 2016, 6, 39520. [Google Scholar] [CrossRef]
- Szabo, G.; Mandrekar, P.; Dolganiuc, A. Innate immune response and hepatic inflammation. Semin. Liver Dis. 2007, 27, 339–350. [Google Scholar] [CrossRef]
- Li, H.; Huang, M.H.; Jiang, J.D.; Peng, Z.G. Hepatitis C: From inflammatory pathogenesis to anti-inflammatory/hepatoprotective therapy. World J. Gastroenterol. 2018, 24, 5297–5311. [Google Scholar] [CrossRef]
- Nakasaki, T.; Tanaka, T.; Okudaira, S.; Hirosawa, M.; Umemoto, E.; Otani, K.; Jin, S.; Bai, Z.; Hayasaka, H.; Fukui, Y.; et al. Involvement of the lysophosphatidic acid-generating enzyme autotaxin in lymphocyte-endothelial cell interactions. Am. J. Pathol. 2008, 173, 1566–1576. [Google Scholar] [CrossRef]
- Zheng, Y.; Voice, J.K.; Kong, Y.; Goetzl, E.J. Altered expression and functional profile of lysophosphatidic acid receptors in mitogen-activated human blood T lymphocytes. FASEB J. 2000, 14, 2387–2389. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Kong, Y.; Mei, B. Lysophosphatidic acid and sphingosine 1-phosphate protection of T cells from apoptosis in association with suppression of Bax. J. Immunol. 1999, 162, 2049–2056. [Google Scholar]
- Gustin, C.; Van Steenbrugge, M.; Raes, M. LPA modulates monocyte migration directly and via LPA-stimulated endothelial cells. Am. J. Physiol. Cell Physiol. 2008, 295, C905–C914. [Google Scholar] [CrossRef]
- Rizza, C.; Leitinger, N.; Yue, J.; Fischer, D.J.; Wang, D.A.; Shih, P.T.; Lee, H.; Tigyi, G.; Berliner, J.A. Lysophosphatidic acid as a regulator of endothelial/leukocyte interaction. Lab. Investig. 1999, 79, 1227–1235. [Google Scholar]
- Schlatzer, D.M.; Sugalski, J.M.; Chen, Y.; Barnholtz-Sloan, J.; Davitkov, P.; Hazlett, F.E.; Funderburg, N.; Rodriguez, B.; Lederman, M.M.; Sieg, S.F.; et al. Plasma proteome analysis reveals overlapping, yet distinct mechanisms of immune activation in chronic HCV and HIV infections. J. Acquir. Immune Defic. Syndr. 2013, 63, 563–571. [Google Scholar] [CrossRef]
- Panther, E.; Idzko, M.; Corinti, S.; Ferrari, D.; Herouy, Y.; Mockenhaupt, M.; Dichmann, S.; Gebicke-Haerter, P.; Di Virgilio, F.; Girolomoni, G.; et al. The influence of lysophosphatidic acid on the functions of human dendritic cells. J. Immunol. 2002, 169, 4129–4135. [Google Scholar] [CrossRef]
- Chen, R.; Roman, J.; Guo, J.; West, E.; McDyer, J.; Williams, M.A.; Georas, S.N. Lysophosphatidic acid modulates the activation of human monocyte-derived dendritic cells. Stem. Cells Dev. 2006, 15, 797–804. [Google Scholar] [CrossRef]
- Rancoule, C.; Viaud, M.; Gres, S.; Viguerie, N.; Decaunes, P.; Bouloumie, A.; Langin, D.; Bascands, J.L.; Valet, P.; Saulnier-Blache, J.S. Pro-fibrotic activity of lysophosphatidic acid in adipose tissue: In vivo and in vitro evidence. Biochim. Biophys. Acta 2014, 1841, 88–96. [Google Scholar] [CrossRef]
- Yamazaki, T.; Joshita, S.; Umemura, T.; Usami, Y.; Sugiura, A.; Fujimori, N.; Kimura, T.; Matsumoto, A.; Igarashi, K.; Ota, M.; et al. Changes in serum levels of autotaxin with direct-acting antiviral therapy in patients with chronic hepatitis C. PLoS ONE 2018, 13, e0195632. [Google Scholar] [CrossRef]


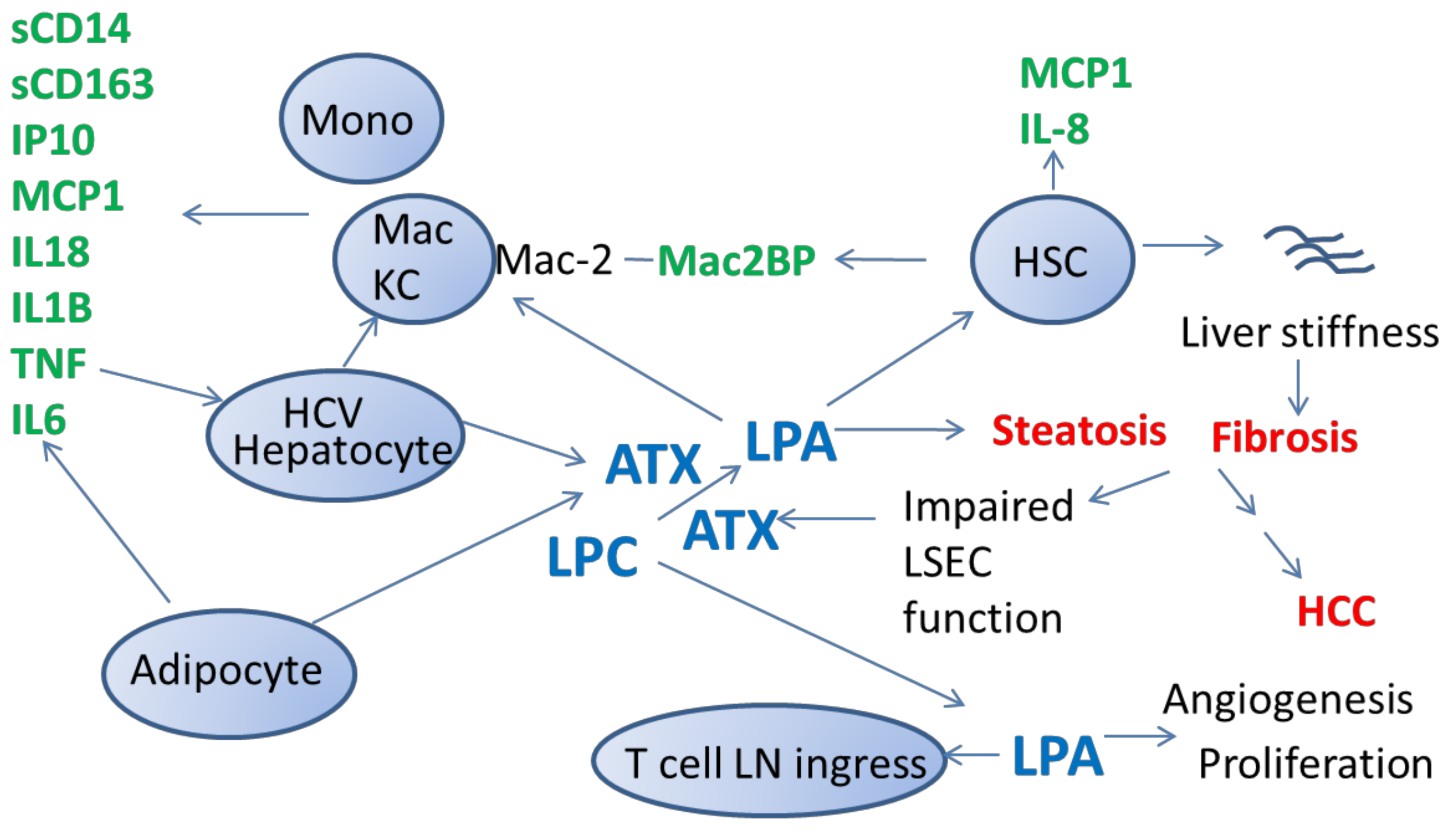{kind=link}
| Study | Disease | ATX Level/Activity | Method of Measurement |
|---|---|---|---|
| Joshita et al. [25] Wunsch et al. [24] | Primary biliary cirrhosis | 0.97 mg/L (0.79–1.11) (serum) 10.2 ± 4.4 nmol/ml/min | 2-site enzyme immunoassay Activity assay method |
| Dhillon et al. [30] Wunsch et al 2016 [24] | Primary Sclerosing Cholangitis | 6.3 nmol/ml/min 7.3 ± 3.4 nmol ml min | Activity assay method Activity assay method |
| Yamazaki et al. [29] Kostadinova et al. [23] Pleli et al. [27] | Chronic HCV infection | 1.39 (1.01–1.99) mg/L (serum) 0.77 mg/L (plasma) 0.814 ± 0.42 mg/L (serum) | 2-site enzyme immunoassay, ELISA ELISA |
| Joshita et al. [22] | Chronic HBV infection | 1.22 mg/L (serum) | 2-site enzyme immunoassay |
| Fujimori et al. [22] Honda et al. [31] Rachakonda et al. [28] | NAFLD1 | 0.86 mg/L (serum) 0.298 mg/L (serum) 0.374 mg/l (serum) | 2-site enzyme immunoassay 2-site enzyme immunoassay ELISA |
| Kondo et al. [32] Enooku et al. [33] | Hepatocellular carcinoma (HCC) | 2.21 ± 1.03 mg/L serum 1.068 mg/L serum | 2-site enzyme immunoassay 2-site enzyme immunoassay |
| Nakamura et al. [34] Pleli et al. [27] Fujimori et al. [22] Kostadinova et al. [23] Wunsch et al [24] | Healthy controls | 0.731 ± 0.176 mg/L serum 0.258 ± 0.40 mg/L (serum) 0.76 mg/L (serum) 0.4 mg/L (plasma) 2.8 ± 1.4 nmol/ml/min | 2-site enzyme immunoassay ELISA 2site enzyme immunoassay ELISA Activity assay method |
| Cytokine or Soluble Receptor | Patient Group | Morbidity | Mortality | Reference |
|---|---|---|---|---|
| IL-6 | Elderly | X | [54,57] | |
| Elderly | Osteoporosis, Alzheimer’s disease, neoplasia, frailty | [56] | ||
| HIV | X | [52] | ||
| HIV | Non-AIDS-defining events: myocardial infarction, stroke, malignancies, serious bacterial infection | [53] | ||
| sCD14 | Elderly | CVD (carotid wall thickness, ankle-brachial index) | X | [61] |
| HCV | Hepatic inflammation, liver fibrosis | [58] | ||
| sCD163 | HCV | Liver fibrosis | [62,63] | |
| HCV/HIV coinfection | Hepatic damage (AST, ALT) | [65] | ||
| HCV/HIV coinfection | Hepatic fibrosis (necroinflammation, Ishak fibrosis score, non-invasive fibrous score) | [64] | ||
| HIV | Non-calcified coronary plaque | [68] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostadinova, L.; Shive, C.L.; Anthony, D.D. Elevated Autotaxin and LPA Levels during Chronic Viral Hepatitis and Hepatocellular Carcinoma Associate with Systemic Immune Activation. Cancers 2019, 11, 1867. https://doi.org/10.3390/cancers11121867
Kostadinova L, Shive CL, Anthony DD. Elevated Autotaxin and LPA Levels during Chronic Viral Hepatitis and Hepatocellular Carcinoma Associate with Systemic Immune Activation. Cancers. 2019; 11(12):1867. https://doi.org/10.3390/cancers11121867
Chicago/Turabian StyleKostadinova, Lenche, Carey L Shive, and Donald D Anthony. 2019. "Elevated Autotaxin and LPA Levels during Chronic Viral Hepatitis and Hepatocellular Carcinoma Associate with Systemic Immune Activation" Cancers 11, no. 12: 1867. https://doi.org/10.3390/cancers11121867
APA StyleKostadinova, L., Shive, C. L., & Anthony, D. D. (2019). Elevated Autotaxin and LPA Levels during Chronic Viral Hepatitis and Hepatocellular Carcinoma Associate with Systemic Immune Activation. Cancers, 11(12), 1867. https://doi.org/10.3390/cancers11121867